Glossary of terms used in theoretical
organic chemistry
[A] [B]
[C] [D] [E]
[F] [G] [H]
[I] [J-K] [L]
[M]
[N] [O] [P]
[Q-R] [S] [T]
[U-V] [W-Z]
H
Half-electron (HE) model -
An approach to treating open-shell molecular systems by a closed-shell
formalism which utilizes the similarity between the SCF equations
(see Hartee-Fock method)
and those for a fictitious closed-shell system in which the odd electron
is replaced by two half- electrons. The model is applied for calculating
energies of radicals by use of
semiempirical quantum
mechanical methods. DEWAR,
HASHNALL, and VENIER (1968).
Hamiltonian - A differential operator
of the total energy. For any
normalized wavefunction y,
the energy is the expectation value of the Hamiltonian operator.
E = 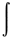 y*Hy
dt
y*Hy
dt
Hard and Soft Acid and Base (HSAB) principle
- A structure-reactivity concept formulated in terms of acid-base
interaction. According to this principle soft acids react faster and
form stronger bonds with soft bases, whereas hard acids react faster
and form stronger bonds with hard bases, everything else being assumed
aproximately equal. Hardness is associated with low polarizability,
high electronegativity
and energy low-lying HOMO (bases) or energy high-lying LUMO (acids).
Softness is related to high polarizability, low electronegativity,
energy high-lying HOMO (bases) and energy low lying LUMO (acids).
The soft-soft preference is characteristic mostly of the reactions controlled
by the orbital interaction,
and the hard- hard preference relates to reactions in which electrostatic
factors prevail. PEARSON (1963); PEARSON
(1990); CHATTARAJ and SCHLEYER (1994).
See also Absolute hardness.
Harmonic approximation
- The approximation of the full nuclear potential of a molecular system
in its equilibrium geometry
and in the vicinity of the respective minimum on the
potential energy surface by the function
V = (1/2) 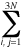 �2V/�qi
�qj
�2V/�qi
�qj
where qi are mass-weighted cartesian displacements
of nuclei relative to their equilibrium positions. The approximation
allows one to describe vibrational motion in terms of independent vibrational
modes normal modes) each of which is governed by a simple one-dimensional
harmonic potential.
Hartree-Fock limit - The lowest
energy that would be obtained via the SCF procedure (see Hartree-Fock
method) if there were no restrictions on the sorts of function
that molecular orbitals could
adopt.
Hartree-Fock (Self-Consistent
Field, SCF) method - Method for determination of the spatial
orbitals yi of the many-electron
determinantal wavefunction (see Slater
determinant) based on reducing coupled nonlinear differential
equations for the optimum forms of the molecular
orbitals by use of the variational method (see variational
principle). The Hartree-Fock hamiltonian
operator is defined in terms of these orbitals through the operators
of coulomb and exchange
repulsion. The general procedure for solving the Hartree-Fock
equations is to make the orbitals self-consistent with the potential
field they generate. It is achieved through an iterative trial-and-error
computational process, for which reason the entire procedure is called
the self-consistent field
method. In the case of open-shell
systems one should distinguish between the spin-restricted
Hartree-Fock (RHF) method and spin-unrestricted
Hartree-Fock (UHF) method. In the former approach a single
set of molecular orbitals is preset, some being doubly occupied
and some being singly occupied with an electron of spin. In the UHF
approach different spatial orbitals are assigned to electrons with
a and b spins
and the orbitals yi doubly occupied
in the RHF method are replaced by two distinct orbitals yi(a)
and yi(b).
HEHRE, RADOM, SCHLEYER, and POPLE (1985).
Heat of atomization - The
negative value of the heat of formation.
It is equal to the total bond energy
of a compound.
Heat of formation - The heat
absorbed or released upon formation of a compound from the elements
in their standard states (i.e. the most stable form of the elements
under standard temperature and pressure, 25oC and 1 atm).
Hessian matrix (synonymous with
force constant matrix) - The matrix of second derivatives of energy.
At any local minimum, all eigenvalues of the Hessian matrix are positive.
At a first-order saddle point, only one eigenvalue is negative and all
others are positive.
Heterolytic bond cleavage - A
bond-dissociation reaction of the type R-X  R-+
X+ resulting in the splitting of the electron pair of
the ruptured bond in a way which leaves the pair with one of the fragments
formed upon dissociation.
R-+
X+ resulting in the splitting of the electron pair of
the ruptured bond in a way which leaves the pair with one of the fragments
formed upon dissociation.
High-spin state - When the separation
between highest occupied and
lowest unoccupied molecular
orbitals is not large,
two alternative electronic states
may be considered for the two uppermost electrons. The state with
the largest number of unpaired electrons is called the high spin
state. In the cases where the one-electron energy needed to promote
an electron to the LUMO is smaller than the Coulomb
and exchange repulsion
energies required to pair up two electrons in the HOMO, the high-spin
state is the ground state.
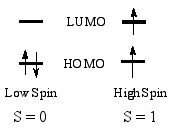
For compounds with more than two unpaired electrons, several spin
states may exist, but in octahedral (or tetrahedral) transition
metal complexes (e.g., organometallic compounds), the high degeneracy
of the MOs results in only one low-spin and one high-spin state.
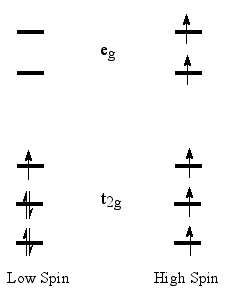
Whether a given compound is in its low-spin
or high-spin state
can be usually determined from magnetic susceptibility or ESR experiments,
although in some cases both states have similar energies and are
thermally populated, giving rise to a peculiar temperature dependence
of the compound’s magnetic behavior.
See also Hund’s rule.
Highest Occupied Molecular Orbital
(HOMO) - see Frontier orbitals.
Homoaromaticity - The aromatic
stabilization of cyclic conjugated systems with 4n + 2
p-electrons is partly preserved when one or more methylene
(or other saturated) groups are inserted into the ring. The molecules
and ions thus formed are regarded as homoaromatic structures. Examples
are cyclobutenyl 1 and cyclooctatrienyl (homotropylium) 2
cations. Trishomocyclopropenyl cation 3 represents a
trishomoaromatic structure.
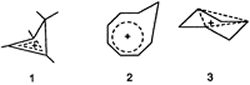
Homodesmotic reaction -
A subclass of isodesmic reactions
in which reactants and products contain equal numbers of carbon
atoms in corresponding states of hybridization;
moreover, there is matching of the carbon-hydrogen bonds in terms
of the number of hydrogen atoms joined to the individual carbon
atoms. To achieve all this matching, one should significantly extend
the number and types of reference molecules. In the aliphatic series
of hydrocarbons these are propane, isobutane and neopentanes as
well as propene and isobutene; for aromatics - buta-1,3-diene, 3-methylidenepenta-1,4-diene
(2-vinylbutadiene) and 3,4-bismethylidenehexa-1,5-diene (2,3-divinylbutadiene).
Thus to assess strain energy
of cyclopropane and aromatic stabilization of benzene the following
homodesmotic reactions are to be respectively analyzed .
c-(CH2)3 + 3 CH3-CH3
3 CH3CH2CH3
DH0exp = -26.5
kcal/mol (110.9 kJ/mol)
C6H6 + 3 CH2=CH2
3 CH2=CH-CH=CH2 (trans)
DH0calc (MP2/6-31G**)
= 23.9 kcal/mol (100.0 kJ/mol)
Due to closer matching of the hybridization states of the atoms
of reactants and products as compared to isodesmic reactions, the
homodesmotic reactions give more accurate estimates of the intrinsic
strain and the cyclic delocalization. The definition may be extended
to molecules with heteroatoms. GEORGE,
TRACHTMAN, BOCK, and BRETT (1975).
Homolytic bond cleavage - The bond-dissociation
reaction of the type R - X R.
+ X. resulting in the splitting of the electron pair
of the ruptured bond in a way that leaves by one electron on each of
the fragments formed upon dissociation.
Hot ground state photoreaction - A
photochemical diabatic reaction
which occurs as the jumps from the potential
energy surface of the excited
state of a molecule to the ground
state surface with excess of thermal energy, large enough
to overcome the potential energy barrier between the reactant and
the product.
Hot state reaction - A reaction proceeding from
an ensemble of molecular entities possessing a higher average vibrational,
rotational or translation energy than they would at thermal equilibrium
with the surrounding medium. IUPAC PHOTOCHEMICAL
GLOSSARY (1988).
See also Hot ground state photoreaction.
Hückel molecular orbital (HMO)
theory - The simplest molecular
orbital theory of p-conjugated molecular
systems. It uses the following approximations: p-electron
approximation; LCAO representation of the
p-molecular orbitals; neglect of electron-electron and nuclear-nuclear
repulsions (in fact, the assumption that these cancel). The diagonal
elements of the effective
Hamiltonian, coulombic
integrals, and the off-diagonal elements,
resonance integrals, (accounted for only directly bonded
atoms) are chosen as empirical parameters, all overlap
integrals being neglected. HEILBRONNER
and BOCK (1976); HÜCKEL (1931).
Hückel resonance energy
- see Resonance energy, various
type of.
Hückel rule - (also known as
the 4n + 2 rule). The electron-counting
rule which states that those monocyclic p-conjugated
molecules and ions are stable which contain (4n + 2)
p-electrons, where n = 0, 1, 2.. .While being originally
formulated on the basis of the simple Hückel
molecular orbital theory, the rule is not limited by the
conditions of this theory and proved to be valid also in the framework
of the self-consistent-field
approximation with electron
correlation taken into account. From the structural point
of view, the rule has certain limitations when applied to polycyclic
systems and heterocycles with highly electronegative or highly electropositive
heteroatoms.
Hund's rule - A rule for building up
the electronic configuration
of atoms and molecules: where a species possesses degenerate orbitals
one electron is placed into each of these before two electrons are
placed in any one of the degenerate set, and the electrons in the singly
occupied orbitals have parallel spins. Thus, the ground
state electron configuration of the nitrogen atom is represented
by N(1s22s22px2py2pz).
Apart from this rule (called sometimes Hund's multiplicity rule), two
other rules of determining ground electronic
states from the magnitudes of the total orbital angular momentum
are referred to as Hund rules. IUPAC PHOTOCHEMICAL
GLOSSARY (1988); BORDEN, IWAMURA, and
BERSON (1994).
Hybrid orbital - An atomic
orbital derived through hybridization
of atomic orbitals with different angular momentum quantum numbers
located at a given atom.
Hybrid Quantum Mechanics/Molecular
Mechanics (QM/MM) methods - A procedure for the teratment of large
molecular systems in such a way that part of the system is treated explicitly
by quantum mechanical (QM) techniques, while the rest of the system
is approximated by a classical or molecular mechanics (MM) force
field. GAO (1998), TOMASI
and POMELLI (1998).
Hybridization - The vectorial type
mixing of atomic orbitals
with different angular momentum quantum numbers on the same atomic
center. Mathematically, the hybridization corresponds to an orthogonal
transformation of a given basis set (e.g. c1=2s,
c2=2px, c3=2py,
c4=2pz) to an
equivalent basis set {ll}.
In simple cases, it may involve the mixing of two (2s, 2px),
three (2s, 2px, 2py) and four (2s,
2px, 2py, 2pz)
AOs, the respective types of hybrid orbitals being referred
to as sp, sp2 and sp3 AOs. For inequivalent ligands
or unequal bond lengths more
general spl hybrid orbitals
have the form:
hl(q)
= N (s + l1/2pq)
where pq is a normalized
p orbital pointing in the direction q, N
is a normalization constant, and l is
generally noninteger.
Hydrogen bond - A particular type
of multicenter (three
center - four electron bond) X-H ...Y in which the central hydrogen
atom covalently linked to an electronegative atom X (C,N,O,S...) forms
an additional weaker bond with atom Y (N,O,S...) in the direction of
its lone electron pair orbital. The energy of hydrogen bonds, which
is usually in the range of 3 - 15 kcal/mol (12 - 65 kJ/mol), results
from the electrostatic interaction and also from the orbital
interaction of the antibonding
s*(XH) MO of the molecule acting
as the hydrogen donor and the nonbonding
lone electron pair MO nY of the hydrogen acceptor
molecule. KOLLMAN and ALLEN (1972);
PIMENTEL and McCLELLAN (1960); PERRIN
and NIELSON (1997).
Hydrophobic effect - The tendency
of nonpolar species to aggregate in water solution so as to decrease
the hydrocarbon - water interfacial area. The effect is a principal
factor determining the structures of proteins and nucleic acids,
the binding of substrates to enzymes, and the binding of antigens
to antibodies. BRESLOW (1991).
Hyperconjugation - The effect
of donation of electron density from the symmetry adapted s-orbitals
of saturated groups to vacant p*-orbitals
of conjugated fragments of a molecular entity; this builds p-character
into bonds that nominally possess only s-character.
See also Negative hyperconjugation.
Hypercoordination - A property
of main-group atoms in molecular entities to acquire
coordination numbers greater than four (which
would comply with the Lewis octet rule).
Hypercoordination may be associated with hypervalency,
but usually is referred to peculiar atomic centers in the electron-deficient
species with multicenter s-bonding, in
which the bonding power of a pair of electrons is spread over more
than two atoms. An example of a hypercoordinated atom is the five-coordinate
carbon atom in the cation methanium, where three C-H bonds may be
regarded as normal two center - two electron bonds and the bonding in
the remaining CH2 fragment is governed by the three-center,
two-electron bond.
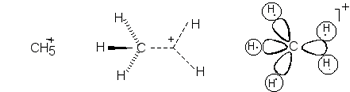
A particular case of a hypercoordinated atom is the hydrogen
atom included into a hydrogen bond. OLAH,
PRAKASH, WILLIAMS, FIELD, and WADE (1987)
Hyperfine coupling - The interaction
between the spin magnetic moment of an unpaired electron and the
nuclear spin magnetic moments resulting in the splitting of the a
(spin up) and b (spin down) energy levels
in an external magnetic field and, thus, in the multiplet pattern of
the ESR spectra of radical-like species and transition metal compounds.
Two main contributions to the hyperfine coupling are usually considered,
Fermi contact and dipolar interactions. The contact interaction
is isotropic and related to the unpaired spin
density at the nucleus, |Yo|2.
The dipolar interaction is anisotropic, and related to r-3,
where r is the distance between the atom holding the unpaired
electron and the nucleus with non-zero spin.
Hyperpolarizability (of nth order)
- The energy of a molecule in an external electrostatic field can be
expanded as
E = Eo - miFi
- (1/2)aijFiFj
- (1/6)bijkFiFjFk
- (1/24)gijklFiFjFkFl
- ...
where Eo is the unperturbed energy, Fi
is the component of the field in the i direction, mi
is the permanent dipole moment,
aij is the polarizability
tensor, and bijk and gijkl
are the first and second hyperpolarizability tensors, respectively.
b is a third order symmetric tensor that measures the second
order response of the molecular electric dipole moment to the action
of an external electric field and thus often referred to as dipole
hyperpolarizability.BÖTTCHER
(1973), HURST, DUPUIS, and CLEMENTI
(1988).
Hypervalency - The ability of an
atom in a molecular entity to expand its valence shell beyond the limits
of the Lewis octet rule. Hypervalent
compounds are common for the second and subsequent row elements
in groups 15-18 of the periodic table. A description of the hypervalent
bonding implies a transfer of the electrons from the central (hypervalent)
atom to the nonbonding molecular
orbitals which it forms with (usually more electronegative)
ligands. A typical example of the hypervalent bond is a linear three-center,
four-electron bond, e.g. that of Fap-P-Fap
fragment of PF5. MUSHER
(1969); REED and SCHLEYER (1990).
