Glossary of terms used in theoretical
organic chemistry
[A] [B]
[C] [D] [E]
[F] [G] [H]
[I] [J-K] [L]
[M]
[N] [O] [P]
[Q-R] [S] [T]
[U-V] [W-Z]
E
ECW model for donor-acceptor interaction
- An approach to obtaining a quantitative scale of bond strengths
of Lewis acid-base complexes. Each acid is characterized by electrostatic,
EA, and covalent, CA, enthalpy
parameters as is each base (EB and CB)
which are combined according to the equation
-DH = EAEB
+ CACB + W
to produce the enthalpy of formation of the AB complex.The E,
C parameters are chosen in such a way as to have minimal
contributions from solvation or lattice energies. The W term
incorporates any constant contribution to the reaction of a particular
acid (or base ) that is independent of the base (acid) it reacts
with. DRAGO, WONG, BILGRIEN, and VOGEL
(1987); VOGEL and DRAGO (1996).
Effective Atomic Number (EAN) rule
- see 18-Electron rule.
Effective Hamiltonian -
A model Hamiltonian, Heff
used for an approximate description of a certain part of the total
electronic system of a molecular entity, e.g. valence electrons or pp-
electrons. It is related to the exact Hamiltonian, H,
for the same problem by means of a factorizing similarity transformation:
Heff = W-1HW
such that Heff has the same eigenvalues as
H (usually for a small subset of the eigenvalues of
H), but much simpler eigenfunctions. In the Hartree-Fock
method Heff
replaces electron-electron repulsion by an average over occupied orbitals.The
Hückel MO theory
can be interpreted as a method based on Heff.
KUTZELNIGG (1988).
see also Kohn-Sham orbitals.
Effective molecular symmetry group
- The appropriate group in which the energy levels of molecules
undergoing rearrangements on a given experimental time scale are to
be classified. The operations of the molecular symmetry group of
such molecules consist of permutations of identical nuclei amongst
themselves, and permutation-inversions, where a permutation is combined
with the inversion of all particle coordinates through the origin
of a space-fixed axis system. The complete nuclear permutation-inversion
group contains all such operations, which all commute with the full
molecular Hamiltonian. LONGUET-HIGGINS
(1963); BUNKER (1979).
See also Symmetry point group,
Symmetry operation.
Electron affinity - The energy
(AX) released upon attachment
of an electron to an atom or a molecule (X) resulting in the formation
of the negative ion X-, i.e.
X + e- 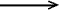 X-
+ AX
X-
+ AX
As with the case of ionization
potential, there may be defined adiabatic
electron affinity and a vertical
electron affinity. The adiabatic AX is equal
to the difference between the total energies of a neutral system
(X) and the corresponding anion (X-). The vertical AX
is equal to the difference between total energies of X and the anion
X- in the equilibrium geometry of X.
Electron correlation - The
adjustment of electron motion to the instantaneous (as opposed to time- averaged)
positions of all the electrons in a molecular system,i. e. the tendency
of electrons to correlate their motions in order to keep as far
apart as possible because of the restrictions set by the Pauli
exclusion principle (exchange correlation) and because of the
electrostatic repulsions (coulombic correlation).
See also Correlation energy.
Electron-counting rules -
Rules establishing correspondence between the topology of a molecular
structure and the number of electrons which may be placed
into its bonding molecular orbitals.
In the origin of various electron-counting rules lies a general assumption
that the completeness of the valence electron shell of a molecular entity
belonging to a certain structural type serves as the major criterion
of structural stability.
See 18-Electron rule, Hückel
rule, Lewis octet rule, Wade's
rules, Woodward-
Hoffmann rules.
Electron deficient compounds -
Molecules or ions that contain too few electrons to allow their
bonding to be described exclusively in terms of two-center, two-electron,
i.e.covalent, bonds. Such molecules or certain fragments
in these are often held together by the multicenter
bonds. The compounds containing atoms with incompletely filled
but spin-paired electron shells (carbenes, carbenium ions) may also
be regarded as electron deficient ones.
Electron density - (synonymous
with charge density), see
Electron density function.
Electron density function
- The electron probability distribution function, r
defined as
r(r) =
n 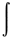 y*[r(1), r(2) ...r(n)]
y [r(1) r(2) ...r(n)]
dr(2)...dr(n)
y*[r(1), r(2) ...r(n)]
y [r(1) r(2) ...r(n)]
dr(2)...dr(n)
where y is an electronic wavefunction
and integration is made over the coordinates of all but the first
electron of n. The physical interpretation of the electron density
function is that rdr gives the
probability of finding an electron in a volume element dr, i.e.
electron density in this volume.
18-Electron rule - for mononuclear
transition metal complexes the electron-counting
rule derived from the fact that transition metals have nine
valence AOs which can be used either for metal-ligand binding or
for accommodating non-bonding electrons. An extension of the rule to
transition metal cluster compounds is known as the effective
atomic number rule.
Electron transfer (ET) reaction
- A redox process in which the overall change that has occurred
is the transfer of one or more electrons.
Electronegativity - The power
of an atom in a molecule to attract electrons. The two widespread empirical
scales of electronegativity are those developed by L. Pauling and R.
Mulliken. The Pauling scale is thermochemical; it is based on the
values of bond energies of type
X-Y, X-X and Y-Y molecules from which the ionic contribution to
the X-Y bond is defined as
DXY = EXY -
(1/2)(EXX + EYY)
From this value the relative electronegativity of X with respect
to Y is defined (in eV1/2 units) as
cX - cY
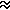 (DXY)1/2
(DXY)1/2
The Mulliken electronegativity ( in eV units) is given by the equation:
cX = (1/2) (IX + AX)
where IX and AX are respectively ionization
potential and electron
affinity in a suitable valence state (see valence
state ionization potential, valence
state electron affinity). both scales are linearly interrelated.
These are useful for estimating bond polarities and strengths of bonds
between different atoms. Many other scales of electronegativity are
known, among which that of A. Allred and E. Rochow , where electronegativity
is defined as the electrostatic force between the nucleus and its
valence electrons, is most frequently used. Accounting for the observation
that the position of bond points
relates to the polarity of a bond, a scale of atomic and group electronegativities,
which are comparable in magnitude to the Pauling values, was derived
(R. Boyd) on the basis of topological properties of the electron
density distributions in model hydrides R-H.
ALLEN (1994); ALLRED and ROCHOW
(1958); BERGMANN and HINZE (1996); BOYD
and BOYD (1992); MULLIKEN (1934);
PAULING (1932).
See also Absolute electronegativity,
Equalization of electronegativity.
Electronic chemical potential
- The quantity that measures the escaping tendency of
electrons from a species in its ground state.
It is the negative of the
absolute electronegativity. PARR,
DONNELLY, LEVY, and PALKE (1978).
Electronic configuration
- The allocation of electrons within an atom or a molecule to a
set of correspondingly atomic
or molecular orbitals
complying with the Pauli exclusion
principle. One electronic configuration may give rise to
several electronic states
with different multiplicities. A wavefunction
for a given electronic
configuration which is an eigenfunction of the electron
spin operators S2 and Sz represents
an electronic state of the the atom or molecule.
Electronic state - An
arrangement allowed by the laws of quantum mechanics of electrons within
an atom, molecule (or system of molecules) .
See also Electronic configuration.
Electronic stability - Unavailability
of another electronic structure (different electronic
state ) of lower energy with the same number of electrons.
Electrostatic potential -
A physical property equal in magnitude to the electrostatic energy
between the static charge distribution, r(r),
of an atomic or molecular system (in the latter case the term molecular
electrostatic potential is commonly used) and a positive unit
point charge located at r. The electrostatic potential V(r)
that is produced at any point r by the electrons and nuclei
(A) of the system is given by
V(r) = 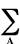 ZA/|RA-rA| -
r(r')dr'/|r'-r|
ZA/|RA-rA| -
r(r')dr'/|r'-r|
POLITZER (1981); SCROCCO
and TOMASI (1973).
Energy gradient - First derivatives
of the total energy with respect to nuclear coordinates, i.e. negative
values of forces on the nuclei. Evaluation of energy gradient plays
a central role in searching potential
energy surfaces for stationary
points. A widespread technique is based on the minimization
of the gradient norm (the
square of energy gradient). SCHLEGEL
(1989).
Energy hypersurface (synonymous
with potential energy surface,
PES) - The notion of hypersurface is used to stress the multidimensionality
of PESs. In a molecular system consisting of N atomic nuclei,
the number of the independent coordinates that fully determine the PES
is equal to 3N-6 (or 3N-5 if the system is linear).
MEZEY (1987).
Energy profile - A schematic plot
of the energy of a reacting system as a function of the reaction
coordinate. The term energy may refer to enthalpy, free
energy or internal energy. Energy profiles are intended to illustrate
the energies of reactant, intermediate, transition and product states
of the system in the order in which they are formed; they are useful
for depicting reaction mechanisms.
Equalization of
electronegativity, principle of - The postulate that in a molecule
all the constituent atoms should have same electronegativity
value, which would be the geometric mean of the electronegativities
of isolated atoms. SANDERSON (1951).
Equilibrium geometry - Molecular
geometry that corresponds to the true minimum on the respective
potential energy surface. While information relating
to the equilibrium geometry is provided by calculations within the
adiabatic approximation (minimization of the total
energy with respect to any independent geometrical parameter),
various experiments yield some effective geometries for the molecule
which are averaged over molecular vibrations.
See also Bond length.
Exchange integral - see Exchange
repulsion.
Exchange repulsion - The correction
to the Coulomb repulsion
between two electrons in orbitals yi
and yj for the case when the electrons
possess parallel spins. It is to be substracted from the Coulomb
repulsion to give the total energy of the electron-electron
interaction. In the Hartree- Fock
theory the magnitude of the exchange repulsion is given by the
exchange integral
Kij = yi*(r1)yj*(r1)(
e2/r12)yi(r2)yj(r2)dr1dr2
= < ij|ji >
For the case of electrons with opposite spins Kij
vanishes.
Excimer - A dimer stable only in the electronically
excited state formed by the interaction
of an excited molecular entity with a ground
state partner of the same structure.
Exciplex - An electronically excited
complex stable only in the electronically excited
state formed by the interaction of an excited molecular
entity with a ground state
partner of a different structure.
Excited configuration -
An electronic configuration
that makes a predominant contribution to the quantum
mechanical description of an excited
state of a system.
Excited state - An electronic
state other than the lowest energy state of a system.
Exciton - A quasiparticle invoked to describe
the migration through the crystal lattice of the excitation of one
molecule in the crystal. The rate of the migration depends on the width
of the band orbital: the wider
bands provide for faster migration.The migration of the excitation is
analogous to the migration through the crystal of a spin-free particle.
FRENKEL (1931); MURRELL
(1963).
Exclusion principle - see
Pauli exclusion principle.
Extended basis set - see
Basis set.
Extended Hückel
MO method (EHMO) - A semiempirical
all-valence electron
quantum mechanical method which uses the same
approximations, apart from p-approximation
and neglect of overlap integrals, as those of the Hückel
molecular orbital theory. The method reproduces relatively
well the shapes and the order of energy levels of molecular
orbitals. The account for overlap makes it possible to describe
the net destabilization caused by interaction of two doubly occupied
orbitals, which effect is not reproduced by HMO theory. HOFFMANN
(1963).
Extended transition state method
(ETS) - An energy partioning scheme of the bond energy DE between
two atoms of fragments A and B into four different terms:
DE (A-B) = DEprep
+ DEelst + DEex
+ DEorb
where DEprep is the energy which
is necessary to promote the fragments A and B from the respective equilibrium
geometry in the electronic ground state
to the geometry and electronic reference state in the molecule A-B. The
three terms DEelst, DEex,
and DEorb give the interaction energy
DEint. They are calculated in three
subsequent steps. DEelst is the
electrostatic interaction energy which is calculated in the first step
with a frozen electron density of the fragments. DEex
then gives the repulsive energy caused by exchange
repulsion, which is calculated when the frozen wavefunction
of step one becomes orthogonalized and antisymmetrized. DEorb
is calculated in the third step. It gives the stabilization which comes
from the orbital interaction
when the wavefunction is completely relaxed. The latter term can be broken
down into orbital contributions with different symmetry. The ETS method
is similar to Morokuma analysis.
ZIEGLER and RAUK (1977).
