Glossary of terms used in theoretical
organic chemistry
[A] [B]
[C] [D] [E]
[F] [G] [H]
[I] [J-K] [L]
[M]
[N] [O] [P]
[Q-R] [S] [T]
[U-V] [W-Z]
B
Back donation (also known as the
Dewar-Chatt-Duncanson model) - A description of the bonding of p- conjugated
ligands to a transition metal which involves a synergic process with
donation of electrons from the filled p-orbital
or lone electron pair orbital of
the ligand into an empty orbital of the metal (donor-acceptor
bond), together with release (back donation) of electrons from an nd
orbital of the metal (which is of p-symmetry
with respect to the metal-ligand axis) into the empty p*-antibonding
orbital of the ligand.
Band orbital - see Crystal
orbital.
Basis function - A one-electron
function used in the expansion of the molecular
orbital function. Basis functions are commonly reperesented
by atomic orbitals (see also
Slater-type orbital or Gaussian-type
orbital) centered on each atom of the molecule.
Basis set - In quantum chemistry, a
set of basis functions employed
for the representation molecular
orbitals. One may distinguish the minimal
basis set (includes one basis function for each SCF occupied
atomic orbital with distinct principal and angular momentum quantum
numbers); split valence basis set
(includes two or more sizes of basis function for each valence orbital);
double zeta DZ) basis set (a
split valence basis set that includes exactly twice as many functions
as the minimal basis set); extended
basis set (the set larger than the double zeta basis set); polarized
basis set (incorporates basis functions of higher angular quantum
number beyond what is required by the atom in its electronic ground
state; allows orbitals to change not only a size, but also a
shape); basis set with diffuse
functions and others. DAVIDSON and
FELLER (1986); FORESMAN and FRISCH
(1996); HEHRE, RADOM, SCHLEYER, and
POPLE (1986); SCHAEFER (1972).
Basis set superposition error
(BSSE) - An artfactual increase in calculated stability of the supersystem
(the system formed by noncovalent interaction between two or more molecular
entities, e.g. hydrogen
bond system) resulting from
the basis set of the supersystem being
larger than for the component subsystems. The BSSE arises from a lowering
of the quantum mechanical energy when the electron
density of each subsystem spreads into the basis functions provided
by the other subsystems. DAVIDSON and FELLER
(1986).
Bent's rule - In a molecule, smaller
bond angles are formed between electronegative ligands since the
central atom, to which the ligands are attached, tends to direct bonding
hybrid orbitals of greater
p character towards its more electronegative substituents. BENT
(1961).
Berry pseudorotation - A
mechanism for the interconversion of trigonal bipyramid structures (1a
and 1b ) through an intermediate (or
transition state ) tetragonal pyramid structure 2.
It may be visualized as two synchronized bending motions by which
a pair of apical ligands (1 and 2) exchange their positions with a pair
of equatorial ones (3 and 4), whereas one equatorial ligand (5)
described as "pivotal" remains unchanged. This process results in an
apparent rotation (pseudorotation) of the actual trigonal bipyramid
structure.
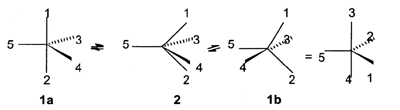
For the majority of five-coordinate main-group and transition
metal compounds the energy difference between the trigonal bipyramid
(1) and tetragonal pyramid (2) structures is sufficiently
low, so that Berry pseudorotation represents a widespread mechanism
of stereochemical nonrigidity.
The same type of intramolecular rearrangement is characteristic also
of four- coordinate bisphenoidal structures, a lone pair always playing
the role of the pivotal phantom-ligand. BERRY
(1960); MISLOW (1970).
See also Pseudorotation.
Bethe lattices - The infinite
connected graphs (a graph is connected
if each pair of its points is joined by a path) not containing cycles,
all vertices of which are equivalent and have equal numbers (n)
of neighbours. Although Bethe lattices with arbitrary n cannot
be realized in three-dimensional space, this model is useful in
the electronic theory of disordered systems.
Bifurcation - Branching of
the minimum energy reaction path
into two different paths at a certain point (bifurcation point)
on the potential energy surface.
Binding energy - The difference
between the total energy of
a molecular system and the sum of the energies of its isolated p-
and s-bonds. The value of binding
energy depends upon the geometrical arrangement of the isolated
subunits (molecules). According to another definition, the term to be
substracted from the total energy is the sum of the energies of
the separate atoms in the corresponding valence states, which compose
the molecule. PAULING (1960).
Biradical - An even-electron molecular
entity with two (in some cases delocalized) radical centers which
act almost independently of each other. The lowest-energy triplet state
of a biradical lies below or at most only a little above its lowest
singlet state.The states of those biradicals whose radical centers
interact particularly weakly are described in terms of a pair of local
doublets. BORDEN (1982); IUPAC
PHOTOCHEMICAL GLOSSARY (1988);
Biradicaloid - A biradical
displaying a strong coupling between the radical centers.
Bloch orbital - see Crystal
orbital.
Bohr magneton - The magnitude of
the standard magnetic moment (the negative first derivative of the
energy with respect to the magnetic field) for an electron:
mB = eh/4pmec
= 0.927408 x 10-23 J T-1 (0.927408
x 10-20 erg G-1)
Bond critical point (synonymous
with bond point) - Within the
topological electron distribution
theory, a (3, -1) critical
point (the point of the gradient field of the electron density within
a given nuclear configuration in which 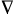 r
(r,q) = 0) which is a local maximum in two directions
and is a local minimum in the third, i.e. a saddle
point in three dimensions. A bond critical point appears
between every pair of neighbouring bonded atoms, its position on the
bond path reflecting the polarity
of a bond. The location of the bond critical point of bond A-B is shifted
toward A and thus, assigns a larger volume of the electronic density
to B if B is more electronegative than A. BADER
(1990).
r
(r,q) = 0) which is a local maximum in two directions
and is a local minimum in the third, i.e. a saddle
point in three dimensions. A bond critical point appears
between every pair of neighbouring bonded atoms, its position on the
bond path reflecting the polarity
of a bond. The location of the bond critical point of bond A-B is shifted
toward A and thus, assigns a larger volume of the electronic density
to B if B is more electronegative than A. BADER
(1990).
Bond ellipticity - Within
topological electron distribution theory, the quantity, e,
which gives a measure of the deviation of the charge distribution
from cylindrical symmetry and thus is correlated with the amount
of p-character of a bond
e = l1/l2
- 1
where l1 and l2
are the principal curvatures of the electron
density function at the bond
point. BADER (1990).
Bond energy - The energy required
to break a given type of bond between atoms in certain valence states.
An averaged bond energy is commonly derived by dissecting the heat
of atomization of a molecule into contributions of individual
bonds. For molecules with localized bonds, the heats of atomization
(formation) are usually well approximated by the sum of pertinent averaged
bond energies. BENSON (1965).
Bond length - The distance
between atomic centers involved in a chemical
bond. The notion of bond length is defined differently in
various experimental methods of determination of molecular geometry;
this leads to small (usually 0.01-0.02 Å) differences in bond
lengths obtained by different techniques. For example, in gas-phase
electron-diffraction experiments, the bond length is the interatomic
distance averaged over all occupied vibrational states at a given temperature.
In an X-ray crystal structural method, the bond length is associated
with the distance between the centroids of electron densities around
the nuclei. In gas-phase microwave spectroscopy, the bond length is
an effective interatomic distance derived from measurements on a
number of isotopic molecules, etc. A number of empirical relationships
between bond lengths and bond orders
in polyatomic molecules were suggested, see, for example, fractional
bond number (the Pauling's bond order).
Bond orbital - A localized
molecular orbital related to a certain s-,
p-, or d-bond.
Bond order - The electron population
in the region between atoms A and B of a molecular entity at the expense
of electron density in
the immediate vicinity of the individual atomic centers. Different schemes
of partitioning electron density give rise to different definitions
of bond orders.In the framework of the Mulliken
population analysis, bond order is associated with the total
overlap population
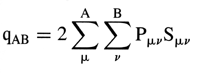
m n where Pmn
and Smn are respectively the elements of
the density matrix and overlap
matrix (see overlap integral).
A large positive value of bond order signifies strong bonding between
the atoms of the molecular entity, whereas negative values of qAB
imply that electrons are displaced away from the interatomic region
and point to an antibonding interaction. In valence
bond theory, bond order is given by a weighted average of
the formal bond orders (i.e. by the number of electron pairs in
a given Lewis structure) between the atoms in the resonance structures
(see Resonance hybrid).
Bond path - Within the topological
electron distribution theory, the line resulting from the addition
of two gradient paths of the electron
density function emanating from the bond
critical point located
between each two neighbouring atomic basins. A bond path can be associated
with all types of interatomic interactions, including hydrogen
bonds and interactions in van
der Waals systems. BADER (1990).
Bond point - see Bond
critical point.
Bond resonance energy (BRE) - A
quantity that represents the contribution of a given p-bond
in a molecule to the topological
resonance energy. p-Bonds with large
negative BREs are presumed to be antiaromatic (see antiaromaticity)
in nature. The greater is the number of such p-bonds
in a molecule, the more the molecule becomes reactive and less thermodynamically
stable. AIHARA (1995).
Bond-atom polarizability
- A quantity used in perturbation
HMO theory as a measure of the change in bond
order, p, between the centers r and s caused
by a change in electronegativity
orcoulomb integral of atom
m:
prs,m = �
prs/� am
Bond-bond polarizability
- A quantity used in perturbation
HMO theory as a measure of the change in bond
order, p, between the centers r and s caused
by a change in the resonance
integral, b, between the
centers m and n:
prs,mn = �
prs/� bmn
Bond-dissociation energy
(BDE) - For a diatomic molecule, the maximum vibrational
energy that a molecule can have prior to its decomposition into
the ground electronic states
of the constituent atoms (spectroscopic bond-dissociation energy,
De). The De value is related to the chemical
dissociation energy: D0 = De - Evib(0),
where Evib(0) is zero-point
vibrational energy. This definition is usually extended
to the dissociation of polyatomic molecules into certain molecular
fragments through homolytic
or heterolytic bond cleavages.
PILAR (1968), FLISZAR
(1994).
Bond-separation reactions
- A class of isodesmic reactions
of considerable importance for the quantitative characterization
of the interaction between neighboring bonds. All formal bonds between
heavy (non-hydrogen) atoms are separated into the simplest reference
(two-heavy atom) molecules containing these same kind linkages.
The set of the molecules involving H, C, N consists of ethane, ethene,
ethyne, methylamine, methanimine, hydrogen cyanide, hydrazine and diazene.
Stoichiometric balance is achieved by the addition of one heavy atom
hydrides (for the H, C, N compounds - methane and ammonia) to the
left-hand side of the reaction scheme. A unique bond-separation
reaction may be drawn for any molecule with a classical valence structure.
The positive bond-separation energy characterizes stabilization
of such a structure with respect to the corresponding isolated bonds
and the opposite is true for negative values of bond-separation energies.
Thus, the bond-separation reactions (1) and (2) reproduce correspondingly
the destabilization energy (strain
energy) of cyclopropane and stabilization energy (due to the
aromaticity) of benzene.
c-(CH2)3 + 3 CH4  3 CH3-CH3 (1)
3 CH3-CH3 (1)
DHexp = -22.1 kcal/mol (-92.5 kJ/mol);
DHcalc(6-31G*) = -26.2 kcal/mol (-110.9
kJ/mol)
C6H6 + 6CH4
3 CH3-CH3 + 3 CH2=CH2 (2)
DHexp = 64.1 kcal/mol (268.2 kJ/mol);
DHcalc(MP2/6-31G*) = 67.2 kcal/mol
(281.2 kJ/mol)
HEHRE, RADOM, SCHLEYER, and POPLE
(1986).
Bond-stretch isomers
- A concept introduced to distinguish molecules (still hypothetical)
which differ only in the length of one or several bonds and correspond
to minima on the same potential
energy surface. A typical
example is given by tricyclo[2.2.2.01,4] octane ([2.2.2]propellane)
in which stretching of the central C-C bond provides for crossing of
the electronic states of
the same symmetry.

By contrast, in experimentally observed spin-state
isomers changes in bond length are associated with changes
in spin state. PARKIN (1993); STOHRER and
HOFFMANN (1972).
Bonding molecular orbital -
A molecular orbital whose
occupation by electrons increases the total bonding (usually, lowers
the total energy) of a molecule. Generally, the energy level of a bonding
MO lies lower than the average of the valence orbitals of the atoms
constituting the molecule.
Born - Oppenheimer
(BO) approximation - Representation of the complete wavefunction
as a product of an electronic and a nuclear part
Y (r,R) = Ye
(r,R)YN(R)
where the two wavefunctions may be determined separately by solving
two different Schroedinger equations. The validity of the Born-Oppenheimer
approximation is founded on the fact that the ratio of electronic
to nuclear mass (m/M @ 5 x
10-4) is sufficiently small and the nuclei, as compared
to the rapidly moving electrons, appear to be fixed.The approximation
breaks down near a point where two electronic
states acquire the same energy (see Jahn-Teller
effect). The BO approximation
m denotes the case when Ye diagonalize
theis often considered as being synonymous with the adiabatic
approximation. More precisely, the latter ter electronic
Hamiltonian. Thus, the adiabatic
approximation is an application of the BO approximation.
Brillouin theorem - The theorem
that states that there is no nonvanishing configuration
interaction matrix elements between the ground-state
determinantal wavefunction and
those determinants resulting from the excitation of one electron
to an empty orbital of the initial SCF calculation
Brillouin zone (usually used as
short for the first Brillouin zone) - The set of all values of the wave
vector which generate non-equivalent crystal
orbitals. It has the form of a polyhedron centered at the
G-point, or the center of the Brillouin zone,
k = (0,0,0). Its component in,e.g.,the x direction
is in the range
- p/a < kx <
p/a
for the first Brillouin zone, where a is the repeat distance
along that direction.
