Glossary of terms used in theoretical
organic chemistry
[A] [B]
[C] [D] [E]
[F] [G] [H]
[I] [J-K] [L]
[M]
[N] [O] [P]
[Q-R] [S] [T]
[U-V] [W-Z]
A
Ab initio quantum mechanical
methods (synonymous with nonempirical
quantum mechanical methods) - Methods of quantum mechanical
calculations independent of any experiment other than the determination
of fundamental observables. The methods are based on the use of the
full Schrödinger equation to treat all the electrons of a chemical
system. In practice, approximations are necessary to restrict the complexity
of the electronic wavefunction
and to make its calculation possible. In this way methods of
density functional theory are usually considered as ab initio
quantum mechanical methods.
Absolute electronegativity
- The property of a chemical system derived from
density functional theory defined as
c = - m = -(�
E/� N)v @
(I + A)/2
where m is the electronic
chemical potential , vis the potential due to the nuclei,
and N is the number of electrons, I and A are respectively
the ionization potential
and electron affinity
of the chemical system in its ground state ( in contrast to a similar
relationship for the Mulliken electronegativity
where I and A refer to the valence state). The "absolute"part
of the term comes from the relationship to the electronic chemical potential,
m . The absolute scale is essentially a measure of the
chemical reactivity of a free atom, molecule, radical or ion, whereas
the Pauling scale of electronegativity
has no meaning with regard to molecules or ions. The scales are,
therefore, comparable only for atoms and radicals where these are roughly
parallel. Absolute electronegativity serves as a measure of bond polarity.
For the species composed of two entities X and Y, the
difference cX- cY
is positive when X-Y has the polarity X-
- Y+. PARR and YANG (1989);
PEARSON (1991).
Absolute hardness -
The resistance of the electronic
chemical potential, m of
a chemical system to a change in the number of electrons as measured
by the curvature of the plot of energy E versus number of
electrons.
h = (1/2)(�
m/� N)v
= (1/2)(� 2E/�
N2)v @ (1/2)(I
-A)
where I and A are respectively ground state
ionization potential
and electron affinity,
and v is the potential due to the nuclei. In molecular
orbital theory, the absolute hardness is measured by the
energy gap between the lowest
unoccupied and highest
occupied molecular orbitals.
h = (eLUMO
- eHOMO)/2
A high value of the absolute hardness is, thus, an
indication of high stability and low reactivity. Absolute
softness is defined as the reciprocal of the hardness. PARR
and YANG (1989); PEARSON (1991).
Absolute softness
- The reciprocal of absolute hardness.
s = 1/h
Active space - Set of
active orbitals in the formalism of Multiconfigurational
SCF method, see also Complete
active space.
Adiabatic approximation
- see Born-Oppenheimer
approximation.
Adiabatic electron
affinity - see Electron
affinity.
Adiabatic ionization
potential - see Ionization
potential.
Adiabatic reaction
- Within the Born-Oppenheimer
approximation, a reaction that occurs on a single potential
energy surface.
Adjacency matrix of
a graph - the matrix which consists of entries aij = 1 for
adjacent vertices, and aij = aii = 0 otherwise. The matrix
is isomorphic to the bonds drawn in simple molecular representation.
Aggregate - An assembly
of molecules stabilized by noncovalent interactions (hydrophobicinteractions,
p-p interactions, ionic
and hydrogen bonds). In contrast
to stable molecules, aggregates are equilibrated mixtures of several
associates corresponding to certain thermodynamic minima. WHITESIDES,
SIMANEK, MATHIAS, SETO, CHIN, MAMMEN and GORDON (1995).
Agostic interaction
- The manner of interaction (termed according to the Greek "to hold
or clasp to oneself as a shield") of a coordinatively unsaturated
metal center with a bond of a ligand. This results in an attraction
between the metal and and the bond and thus often in structural distortions
of the whole complex. Initially described for a C - H -Metal bond
interaction where M is a transition metal complex, it
has been commonly used to describe M...YZ interaction. It is thought
to be determining in the activation of a bond, notably C-H.
BROOKHART and GREEN (1983).
Alternancy symmetry
- A topological property of the molecular
graphs of alternant hydrocarbons
which allows the carbon atoms to be divided into two subsets in
such a way that no two atoms of the same subset are adjacent. A
consequence of this property is the symmetrical arrangement of the
energy levels of bonding
and antibonding Hückel
MOs relative to the level of a nonbonding
orbital (energy level of the p AO of a carbon atom).
Alternant hydrocarbon
- A conjugated hydrocarbon whose molecule does not contain
odd-membered rings, so that it is possible to divide the carbon
atoms into two sets, "starred" atoms and "unstarred" atoms in such
a way that no two atoms of the same set are linked by a bond.
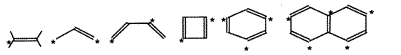
If the total number of starred and unstarred atoms in
an alternant hydrocarbon is even, it is assigned to the even alternant
hydrocarbon type. If this number is odd, the hydrocarbon belongs to
the type of odd alternant hydrocarbons. The molecular
orbitals and energy levels of alternant hydrocarbons are
perfectly paired (see Perfect pairing).
Angular Overlap Model
(AOM) - A method of description of transition metal - ligand interactions
and main-group element stereochemistry, whose basic assumption is
in that the strength of a bond formed using atomic
orbitals on two atoms is related to the magnitude of overlap
of the two orbitals. The interactions between the central-atom and ligand
orbitals are usually divided into the s-,
p- and d-types
and parametric equations of the type
estab, s
= F2es
- (F2)2fs
edestab, s
= - [F2es - (F2)2fs]
are used, where F is angle-dependent contribution
to the overlap integral
Sab between the two interacting orbitals, whereas
parameters es and fs
are proportional to S2 and S4 respectively
and depend on the identity of atoms A and B as well the A - B bond
distance. Similar equations are derived for the p-
and d-type interactions. Neither orbital
mixing nor nuclear repulsions are accounted for by the model. Its
advantage is in that for simple systems a molecular orbital diagram
is easily constructed on the basis of two-orbital interactions and
clearly reveals trends in orbital energies on distortion. BURDETT
(1980); RICHARDSON (1993).
Antiaromaticity (antithetical
to aromaticity) - Those cyclic molecules
for which cyclic electron delocalization provides for the reduction
(in some cases loss) of thermodynamic stability compared to acyclic
structural analogues are classified as antiaromatic species. In contrast
to aromatic compounds, antiaromatic ones are prone to reactions
causing changes in their structural type, and display tendency to alternation
of bond lengths and fluxional behavior (see fluxional
molecules) both in solution and in the solid . Antiaromatic
molecules possess negative (or very low positive) values of resonance
energy and a small energy gap between their highest
occupied and lowest
unoccupied molecular orbitals. In antiaromatic molecules, an
external magnetic field induces a paramagnetic electron current.
Whereas benzene represents the prototypical aromatic compound, cyclobuta-1,3-diene
exemplifies the compound with most clearly defined antiaromatic properties.
BRESLOW (1973); MINKIN,
GLUKHOVTSEV and SIMKIN (1994).
Antibonding molecular
orbital - The molecular orbital
whose occupation by electrons decreases the total bonding (as
usual, increases the total energy) of a molecule. In general, the energy
level of an antibonding MO lies higher than the average of the valence
atomic orbitals of the atoms
constituting the molecule.
Antisymmetry principle
(synonymous with the Pauli exclusion
principle) - The postulate that electrons must be
described by wavefunctions which
are antisymmetric with respect to interchange of the coordinates (including
spin) of a pair of electrons. A corollary of the principle is the Pauli
exclusion principle. All particles with half-integral spin (fermions)
are described by antisymmetric wavefunctions, and all particles
with zero or integral spin (bosons) are described by symmetric wavefunctions.
Apicophilicity - In
trigonal bipyramidal structures with a five-coordinate central atom,
the stabilization achieved through a ligand changing its position
from equatorial to apical (axial). The apicophilicity of an atom or
a group is evaluated by either the energy difference between the stereoisomers
(permutational isomers) containing the ligand in apical and equatorial
positions or the energy barrier to permutational
isomerization (see also Berry
pseudorotation). In general, the greater the electronegativity
and the stronger the p-electron-withdrawing
properties of a ligand (as for Cl, F, CN), the higher is its apicophilicity.
The notion of apicophilicity has been extended to four-coordinate
bisphenoidal and three-coordinate T-shaped structures, which can be
viewed as trigonal bipyramidal species where respectively one or
two vertices are occupied by phantom ligands (lone electron pairs).
TRIPPETT (1974); McDOWELL
and STREITWIESER (1985).
Aromaticity - The concept
of spatial and electronic structure of cyclic molecular systems displaying
the effects of cyclic electron delocalization which provide for
their enhanced thermodynamic stability (relative to acyclic structural
analogues) and tendency to retain the structural type in the course
of chemical transformations. A quantitative assessment of the degree
of aromaticity is given by the value of the resonance
energy. It may also be evaluated by the energies of relevant
isodesmic and
homodesmotic reactions. Along
with energetic criteria of aromaticity, important and complementary
are also a structural criterion (the lesser the alternation of bond
lengths in the rings, the greater is the aromaticity of the molecule)
and a magnetic criterion (existence of the diamagnetic ring current
induced in a conjugated cyclic molecule by an external magnetic field
and manifested by an exaltation and anisotropy of magnetic susceptibility).
Although originally introduced for characterization of peculiar
properties of cyclic conjugated hydrocarbons and their ions, the
concept of aromaticity has been extended to their homoderivatives (see
homoaromaticity), conjugated
heterocyclic compounds (heteroaromaticity), saturated cyclic compounds
(s-aromaticity) as well as to three-dimensional
organic and organometallic compounds (three-dimensional aromaticity).
A common feature of the electronic structure inherent in all aromatic
molecules is the close nature of their valence electron shells, i.e.
double electron occupation of all bonding MOs with all antibonding
and delocalized nonbonding
MOs unfilled. Thenotion of aromaticity is applied also to
transition state. GARRAT
(1986); MINKIN, GLUKHOVTSEV and SIMKIN
(1994), SCHLEYER and JIAO (1996).
See also Electron counting
rules, Hückel rule.
Atom-atom polarizability
- A quantity used in perturbation
HMO theory as a measure of the change in electron density,
q, of atom s caused by a change in the electronegativity
(or coulomb integral),
ar, of atom r:
pSI
= � qs/�
ar
Atom-bond polarizability
- A quantity used in perturbation
HMO theory as a measure of the change in electron density,
q, of atom m caused by a change in the resonance integral,
b, of bond rs:
pm,rs = �
qm/� brs
Atomic basin - Within
the topological electron distribution
theory, the region of three-dimensional space defined by
the gradient paths of the charge density which terminate at each nucleus
in a molecule. The atomic basin is an unambiguous definition of
an atom in a molecule.
Atomic charge - The
charge attributed to an atom A within a molecule defined as
z = ZA - qA, where ZA is the atomic
number of A and qA is the electron density assigned to A. The
method of calculation of qA depends on the choice of the scheme
of partitioning electron density. In the framework of the Mulliken
population analysis qA is associated with the so-called
gross atomic population: 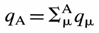 where
qm is a gross population
for an orbital m in the basis set employed
defined according to
where
qm is a gross population
for an orbital m in the basis set employed
defined according to
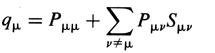
where Pmn and
Smn are the elements of
density matrix and overlap matrix respectively (see
overlap integral). In the Hückel
molecular orbital theory (where Smn
= dmn),
qm= nmPmm
, where nm is the number of
electrons in the MO m. HEHRE,
RADOM, SCHLEYER, and POPLE (1986).
Atomic orbital - see
Orbital.
Atomic units - The units
designed to simplify the form of the fundamental equations of quantum
mechanics by eliminating from them fundamental constants. The atomic
unit of length is the Bohr radius, ao = h2/4p2me2
= 5.291 77249 x 10-11 m (0.529177249 Å).
Energy is measured in hartrees, where 1 hartree = e2/ao
= 4.359 7482 x 10-18 J. Masses are specified in
terms of atomic mass unit, amu = 1.6605402 x 10-27 kg and
of the electron mass unit, me = 0.910953 x 10-30
kg. The advantage of atomic units is that if all calculations are directly
expressed in such units, the results do not vary with any revision of
the numerical values of the fundamental constants. COHEN
and TAYLOR (1986).
Atomization energy
- synonymous with Heat of atomization.
Atoms in molecules
(AIM), theory of - A quantum chemical method based on the
assumption that the wavefunction
of a molecule can be expressed as a linear combination
Y = S
ciYi
where Yi are
the antisymmetrized products of wavefunctions 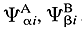 ,
… of atoms A, B… in electronic
states ai, bi
,…BADER (1990); BADER,
POPELIER, and KEITH (1994)
,
… of atoms A, B… in electronic
states ai, bi
,…BADER (1990); BADER,
POPELIER, and KEITH (1994)
See also Topological
electron distribution theory.
Aufbau principle -
A rule for building up the electronic configuration of atoms and molecules.
It states that a maximum of two electrons are put into orbitals
in the order of increasing orbital energy: the lowest-energy orbitals
are filled before electrons are placed in higher-energy orbitals.
See also Pauli exclusion principle
and Hund's rule.
Automerization (synonymous
with degenerate rearrangement,
permutational isomerization,
topomerization) - A molecular
rearrangement in which the reactant is transformed to the product
which differs from reactant only in the permutation of identical atoms.
Automerizations may be detectable by the methods which allow one to
distinguish individual atoms within a molecule: by isotopic labelling
and by dynamic nuclear magnetic resonance technique. An example of an
automerization reaction is the photochemical rearrangement of benzene
via tricyclo[3.1.0.02,6]hex-3-ene (benzvalene).
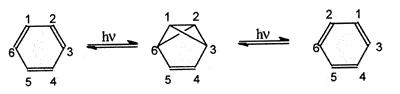
BALABAN and FARCASIU
(1967); BALABAN, GHEORGHIU, SCHIKETANZ,
and NECULA (1989); BINSCH,ELIEL
and KESSLER (1971); MINKIN, OLEKHNOVICH
and ZHDANOV (1988).
Avoided crossing -
Within the Born-Oppenheimer
approximation, when two electronic
states change their energy order as the molecular geometry
is continuously changed along a reaction
path, their energies may become equal at some points (surface
crossing) or only come relatively close (the surface crossing is said
to be avoided). If the electronic states are of the same symmetry, the
surface crossing is always avoided in diatomics and usually avoided
in polyatomics. SALEM (1982)
See also Noncrossing rule.
