Ph.D. Thesis
Title Towards an understanding of simple reactions
in Heterogeneous Catalysis
Adviser Dr Peijun Hu
Thesis Committee Robbie Burch, School of Chemistry, The Queen's
University of Belfast; Richard Catlow, Royal Institution of Great
Britain.
Essay
Few fields of scientific endeavour can have contributed
so dramatically to human existence over the last forty years as heterogeneous
catalysis. Over 80 % of industrial chemical production proceeds with
the aid of a solid catalyst, and heterogeneous catalysis represents
20-30 % of global GNP annually. The key to future advances in heterogeneous
catalysis must be to understand surface processes at an atomic level,
and the surface catalysed reaction is of paramount importance. Aside
from the certain fundamental significance of such an understanding,
it would facilitate the development of designer catalysts with enhanced
reactivities and selectivities.
Acquiring this knowledge is not, however, an easy task.
To determine microscopic reaction pathways high temporal (picosecond)
as well as high spatial (Angstrom) resolution is simultaneously required,
making experimental investigations in this area problematic. Computer
simulations - chemistry in a "virtual laboratory" - are, therefore,
an attractive alternative. Thanks to recent methodological and computational
developments it is now possible to apply highly accurate quantum mechanical
techniques to the study of catalytic reactions. The accuracy of one
particular technique, density functional theory (DFT), has improved
dramatically over recent years with the effect that DFT calculations
now yield precise structural and energetic information. This makes
DFT particularly amenable to the study of reaction pathways and barriers.
My studies involved the application of state-of-the-art
DFT methods to surface catalysed reactions of relevance to heterogeneous
catalysis. Our approach was to model reactions in simulation cells.
Each cell contains 20-30 atoms, repeated infinitely in three dimensions.
By determining the electronic structures of these infinite systems,
accurate descriptions of catalytic surfaces, with arrays of reactants
adsorbed upon them, can be obtained. Simply by reducing the distance
between the reactants and obtaining updated electronic and molecular
structures one can determine both low energy reaction pathways and
their associated activation energies. A variety of reaction systems
have been examined. I will present three examples, each of particular
importance, which illustrate well the sorts of issues that can be
addressed by theoretical chemistry.
A new mechanism for hydrogenation
Hydrogenation is a process of considerable scientific and technological
importance. The conventional view in heterogeneous catalysis had been
that H atoms adsorbed on the surface of the catalyst carry out the
hydrogenation reactions. Recently, however, a group from M.I.T. revealed
an exciting new hydrogenation mechanism. It was demonstrated that
H atoms absorbed within a Ni catalyst (subsurface H atoms) were more
reactive than H atoms adsorbed on the surface of the catalyst (surface
H atoms). It was shown, moreover, that certain hydrogenation reactions
are only possible when subsurface H atoms are involved. It was not
clear, however, how this new class of reaction proceeds or why subsurface
H atoms were so reactive. In the first application of DFT to reactions
involving subsurface species, we determined microscopic reaction pathways
for reactions involving surface and subsurface H atoms (Figure 1).
Quantum mechanics was used to reveal precisely how subsurface H atoms
diffuse and react at the selvedge of a Ni catalyst. By careful examination
of each reaction pathway and barrier a model was proposed to explain
the high reactivity of subsurface H atoms. Essentially the bond strength
between a subsurface H and the Ni catalyst is weaker than that between
a surface H and the Ni catalyst, due to greater nuclei-nuclei repulsion
experienced by subsurface H atoms. As a consequence subsurface H atoms
are metastable with respect to surface H and it is this metastability
that accounts for the observed reactivity. This model is of some importance
since it can be applied generally in this new important class of catalytic
reactions. Furthermore, it will assist in the identification of other
systems in which subsurface species have increased reactivities, thus
facilitating the identification of new catalysts.
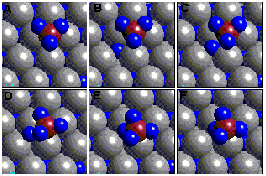
Figure
1. Microscopic reaction pathway for a subsurface H (blue) reacting
with methyl to produce methane on the surface of a Ni (grey) catalyst.
Catalysis: back to the beginning
On contact with platinum, hydrogen and oxygen react to produce water.
The remarkable ease with which this reaction proceeds prompted Berzelius
in 1836 to first use the term "catalysis". Despite apparent simplicity,
its mechanism has remained the subject of much debate. We have performed
an extensive study of this original catalytic process. Two distinct
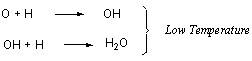
temperature dependent mechanisms of H2O formation have
been identified. At high temperatures H2O is formed by
the simple and intuitively attractive addition of H to O and then
to OH:
At low temperatures a proton transfer reaction (disproportionation
reaction) from H2O to O produces hydroxyls and ultimately
H2O much more readily than the reaction of O and H:
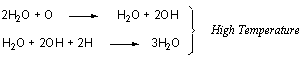
This dual path mechanism to H2O formation
is an important finding, one that nicely explains and rationalises
many seemingly contradictory experimental observations.
A further significant discovery is the role played by
H2O in catalysis. In the low temperature mechanism one
of the H2O molecules dramatically improves the efficacy
of the disproportionation process. Although not directly involved
in the reaction this H2O molecule, through hydrogen-bonding,
lowers the transition and final state energies and acts as an extremely
effective thermodynamic and kinetic promoter. Because of the ubiquitous
presence of H2O molecules in industrial catalytic systems
this effect should be considered in the future design of industrial
catalytic systems.
A new framework
Finally, perhaps the most exciting example of my research is a systematic
study into the microscopic reaction pathways of catalytic reactions.
The basic principles which govern the pathways of most chemical reactions,
especially those of relevance to heterogeneous catalysis, are not
yet clear. Our objective was the development of a generalised framework.
By careful examination of twelve catalytic reaction pathways of our
own determination, and others from the literature, we were able to
make important initial steps towards this goal. Crucially, we discovered
that the valency of the reactants involved is the principal determining
factor of a catalytic reaction pathway. Reactants of the same valency
access similar transition states and the location of transition states
is determined by the valency of the reactants involved. To provide
a simple example CH4, NH3 and H2O,
all saturated molecules, dissociate preferentially over a single metal
atom on a Pt surface. On this basis, we developed rules to facilitate
the prediction of catalytic reaction pathways without recourse to
experimental or theoretical investigations.
Conclusion
The application of modern quantum mechanical techniques to the study
of surface catalysed reactions has been the focus of my work. Studies
such as those outlined reveal how quantum mechanics can be used to
accurately illustrate important issues in heterogeneous catalysis.
Theory facilitates our better understanding of the most intimate details
of catalytic reactions and assists in the translation of this data
into a conceptual form that fellow chemists can use to design new
processes .
