- ... 1961-19711
- The membership of the Commission during this period was as follows:
Chairman: 1961-67 Sir Eric Rideal (UK); 1967-69 J. Th. G. Overbeek (Netherlands); 1969- D. H. Everett (UK);
Secretary: 1961-67 W. A. Zisman (USA); 1967- H. van Olphen (USA);
Titular members: 1961-65 A. E. Alexander (Australia); 1967- S. Brunauer (USA); 1969-. L. Burwell (USA); 1961-65 D. G. Dervichian (France); 1961-69 M. M. Dubinin (USSR); 1965-69 D. H. Everett (UK); 1961-65 K. Groth (Sweden); 1969- R. Haul (Germany); 1961-67 J. Horiuti (Japan); 1961-69 B. Kamienski (Poland); 1969V. V. Kazansky (USSR); 1969- J. Mysels (USA): 1961-67 J. Th. G. Overbeek (Netherlands); 1965-71 M. Prettre (France): 1967- G. Schay (Hungary).
Associate Members: 1969- R. M. Barrer (UK); 1967- G. Boreskov (USSR); 1967-69 R. L. Burwell (USA); 1969- S. Friberg (Sweden); 1967-69 R. Haul (Germany); 1967-71 J. Horiuti (Japan); 1969- C. Kemball (UK); 1969- A. V. Kiselev (USSR); 1969- H. Lange (Germany): 1967-69 K. J. Mysels (USA); 1967- Sir Eric Rideal (UK); 1967-A. Scheludko (Bulgaria); 1969- G. A. Schuit (Netherlands); 1965-1967 H. van Olphen (USA).
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... interface2
- The use of a solidus to separate the names of the bulk phases is preferred to the use of a hyphen which can lead to ambiguities. The same applies to the abbreviated notation for phase boundaries, i.e. S/L: S/G: L/L: L/G: S/L/G
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... molecules3
- The term molecule is used here in the general sense to denote any molecular species : atom, ion, neutral molecule or radical.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... solids4
- (2001) A system of terms applicable to ordered microporous and mesoporous inorganic materials, such as zeolites, can be found in ``Nomenclature of structural and compositional characteristics of ordered microporous and mesoporous materials with inorganic hosts'', McCusker, L.B., Liebau, F., Engelhardt, G., Pure Appl. Chem. 73 (2001), 381-394
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
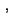 5
5
- (2001) A special class of materials with microporosity and mesoporosity are the pillared layer solids. The terminology used for these materials and their characterization are discussed briefly in ``Pillared clays and pillared layered solids'', Schoonheydt, R.A., Pinnavaia, T., Lagaly, G., Gangas, N., Pure Appl. Chem. 71 (1999) 2367-2371.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... values6
- (2001) Recommendations for the assessment of the porosity can be found in:
(1) Sing, K.S.W., Everett, D.H., Haul, R.A.W., Moscou, L., Pierotti, R.A., Rouquerol, J., Siemieniewska, T., ``Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity'', Pure Appl. Chem. 57 (1985) 603-619 and (2) Rouquerol, J., Avnir, D., Fairbridge, C.W., Everett, D.H., Haynes, J.H., Pernicone, N., Ramsay, J.D.F., Sing, K.S.W., Unger, K.K., ``Recommendations for the characterization of porous solids'', Pure Appl. Chem. 66 (1994) 1739-1758.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... micropores7
- (2001) In micropore filling the whole of the assessable volume present in the micropores is regarded as adsorption space, as distinct from surface coverage which takes place on the walls of macropores or mesopores.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... measurement8
- (2001) Recommendations on the use of gas adsorption for the measurement of the micropore volume, the mesopore volume and capillary condensation can be found in: Sing, K.S.W., Everett, D.H., Haul, R.A.W., Moscou, L., Pierotti, R.A., Rouquerol, J., Siemieniewska, T., Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity'', Pure Appl. Chem. 57 (1985) 603-619.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... menisci9
- (2001) See also previous footnote.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
surface10
- The abbreviated form is generally preferred.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... monolayer11
- (2001) The principles for measuring and reporting experimental data on spread monolayers are discussed in: Ter-Minassian-Saraga, L., ``Reporting experimental pressure-area data with film balances'', Pure Appl. Chem. 57 (1985) 621-632.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... surface)12
- In the field of spread monolayers it has been customary to use
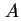 for the area per molecule and
for the area per molecule and 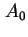 for its co-area (the two-dimensional analogue of the co-volume of a real gas). To avoid confusion with
for its co-area (the two-dimensional analogue of the co-volume of a real gas). To avoid confusion with 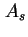 for the total area and for Helmholtz energy, the adoption of
for the total area and for Helmholtz energy, the adoption of 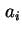 (or
(or 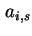 ) and
) and 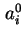 (or
(or 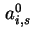 ) for area and co-area per molecule respectively is recommended.
) for area and co-area per molecule respectively is recommended.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... interface13
- (2001) A supplement to the present definitions and the operational determination of adsorption from solution, together with the interpretation of the data in thermodynamic terms is discussed in Everett, D.H., ``Reporting data on adsorption from solution at the solid/solution interface'', Pure Appl. Chem. 58 (1986) 967-984.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...)14
- (2001) The evaluation of gas adsorption isotherms is discussed in: Sing, K.S.W., Everett, D.H., Haul, R.A.W., Moscou, L., Pierotti, R.A. Rouquerol, J., Siemieniewska, T., ``Reporting physisorption data for gas/solid systems with special reference to the determinations of surface area and porosity'', Pure Appl. Chem. 57 (1985) 603-619.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... methods15
- (2001) Recommendations on the determination of specific surface areas by application of the BET-method to gas adsorption data are presented in: Sing et al. Pure Appl. Chem. 57 (1985) 603-619.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...)16
- The use of the term
adhesion tension is discouraged because it can be
confused with the term work of adhesion (see above).
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... rheology17
- (2001) A set of terms of particular interest to colloid and surface chemists can be found in: Lyklema, J. and van Olphen, H., ``Terminology and symbols in colloid and surface chemistry - part 1.13: definitions, terminology and symbols for rheological properties'', Pure Appl. Chem. 51 (1979) 1213-1218.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... by18
- Mass is employed as extensive variable for the adsorbent in these equations because the area may change on adsorption.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...)19
- Formerly called the isosteric heat of adsorption.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... as20
- The naming of these quantities as enthalpies and their notation by
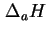 is not strictly justified since
is not strictly justified since
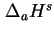 is a difference between two differently defined enthalpies, and
is a difference between two differently defined enthalpies, and
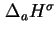 is the difference between an energy and an enthalpy. A more complicated notation, which is hardly justified, would be needed to indicate these distinctions.
is the difference between an energy and an enthalpy. A more complicated notation, which is hardly justified, would be needed to indicate these distinctions.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... chosen21
- (2001) The superscript * is recommended for reference to a pure liquid.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... group22
- This assumes that one of the two phases is aqueous and the other non-aqueous. If both are non-aqueous (e.g. oil/air) molecules containing organophilic and organophobic groups may be amphipathic and surface active.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... stated23
- (2001) See also: K.J. Mysels, P. Mujerjee, ``Reporting experimental data dealing with critical micellization concentrations (c.m.c.'s) of aqueous surfactant systems'', Pure Appl. Chem. 51 (1979): 1083-1089.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... constant)24
- (2001) In the original text the symbol is used for the van der Waals-Hamaker constant. The present notation is recommended in the ``Green Book''.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... (20\celsius)25
- It would be preferable to call this quantity the standard sedimentation coefficient.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... Electrochemistry26
- (2001) The definitions and terminology most relevant for colloid and surface chemists can be found in: Parsons, R., ``Manual of Symbols and Terminology for Physicochemical Quantities and Units: Appendix III: Electrochemical nomenclature'', Pure Appl. Chem. 34 (1974) 500-516.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
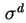 27
27
- (2001) In the original document, the symbols
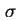 ,
,
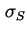 and
and
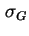 are used for respectively the charge densities at the surface plane, in the Stern layer and in the Gouy or diffuse layer. However, these notations are no longer recommended. The present notation is in accordance with the recommendations by (1) Parsons, R., ``Manual of Symbols and Terminology for Physicochemical Quantities and Units: Appendix III: Electrochemical nomenclature'', Pure Appl. Chem. 34 (1974) 500-516 and (2) Lyklema, J., ``Electrified interfaces in aqueous dispersions of solid, Pure Appl. Chem. 63 (1991) 895-906.
are used for respectively the charge densities at the surface plane, in the Stern layer and in the Gouy or diffuse layer. However, these notations are no longer recommended. The present notation is in accordance with the recommendations by (1) Parsons, R., ``Manual of Symbols and Terminology for Physicochemical Quantities and Units: Appendix III: Electrochemical nomenclature'', Pure Appl. Chem. 34 (1974) 500-516 and (2) Lyklema, J., ``Electrified interfaces in aqueous dispersions of solid, Pure Appl. Chem. 63 (1991) 895-906.
The inner boundary of the diffuse layer is called the Stern plane or outer Helmholtz plane. The charge
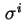 in the Stern layer is generally assumed to be located at the inner Helmholtz plane, this is a plane situated between the surface plane and the Stern plane.
in the Stern layer is generally assumed to be located at the inner Helmholtz plane, this is a plane situated between the surface plane and the Stern plane.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
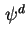 28
28
- (2001) In the original document, the electrical potential is indicated as inner or Galvani potential,
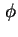 . For a solid phase immersed in a liquid phase this is the potential difference between a point in the bulk phase of the solid and a reference point
. For a solid phase immersed in a liquid phase this is the potential difference between a point in the bulk phase of the solid and a reference point 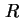 beyond the electric field of the solid in the liquid phase. The outer or Volta potential,
beyond the electric field of the solid in the liquid phase. The outer or Volta potential, 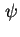 , for this situation is the potential difference just outside the solid phase and the reference point in the liquid phase. The difference between and is called the surface electric potential or ``
, for this situation is the potential difference just outside the solid phase and the reference point in the liquid phase. The difference between and is called the surface electric potential or ``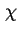 '' potential.
'' potential.
If it is assumed that the surface potential is the potential at the outer boundary of the solid, no phase boundary is crossed between the surface and the reference point . Under these conditions the surface potential can be considered as an outer potential . Potentials inside the diffuse double layer are also defined with respect to point in the liquid phase and these are outer potentials. Therefore, in colloid chemistry the surface potential and the potentials in the double layer are mostly all denoted as outer potentials, .
In accordance with the distinctions of the double layer charges a distinction can be made between the surface potential, 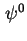 , the potential at the inner Helmholtz plane,
, the potential at the inner Helmholtz plane, 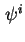 , and the potential at the Stern layer (the inner boundary of the diffuse layer), . The course of the potential as a function of the distance to the interface is denoted as
, and the potential at the Stern layer (the inner boundary of the diffuse layer), . The course of the potential as a function of the distance to the interface is denoted as 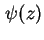 , or
, or 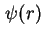 where
where 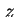 or
or 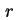 is the distance to the surface. These potentials cannot be measured directly. In most cases a double layer model is used to convert the various charge densities into the corresponding potentials.
is the distance to the surface. These potentials cannot be measured directly. In most cases a double layer model is used to convert the various charge densities into the corresponding potentials.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...29
- (2001) In the original text the symbol
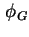 is used for the potential at the inner boundary of the diffuse layer, this symbol is not in accordance with more recent recommendations.
is used for the potential at the inner boundary of the diffuse layer, this symbol is not in accordance with more recent recommendations.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
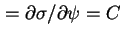 dl30
dl30
- (2001) In the original text the symbols
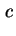 dl and
dl and
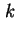 dl are used, according to recent rules, capitals are preferred.
dl are used, according to recent rules, capitals are preferred.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
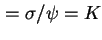 dl31
dl31
- (2001) See previous footnote.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... (i.e.p.)32
- (2001) The difference between the p.z.c. and the i.e.p. is further explained by Lyklema, Pure Appl. Chem. 63 (1991) 895-906.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... ions33
- (2001) As pointed out by Lyklema, Pure Appl. Chem. 63 (1991) 895-906, for disperse systems the surface charge density is the primary parameter rather than the surface potential. The term charge determining ions is therefore in most cases more appropriate.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
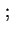 34
34
- (2001) Presently, the rationalized four-quantity system (using S.I. units) is strongly recommended
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...ELECTROKINETICS35
- To be distinguished from electrode kinetics.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... recommended36
- (2001) A good example of constistent usage of modern notation can be found in J. Lyklema, Fundamentals of Interface and Colloid Science, Vol II, Academic Press (1995), Chapter 4.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...)37
- (2001) Older units should be avoided. To avoid confusion, it is wise to indicate which units are used in each particular case.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
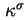 38
38
- (2001) The surface (excess) conductivity can have contributions due to the diffuse layer charge outside the plane of shear (also called ``Bikerman'' surface conductivity) and due to the charge behind the plane of shear in the stagnant layer (stagnant layer conductivity).
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... plane39
- (2001) The shear plane is also called slip(ping) plane. The
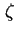 potential is identified as the potential at the supposed slipping plane that separates the stationary and the mobile phases in tangential flow of the liquid with respect to the surface. Although there is still very little direct evidence to support the view of a slipping plane (instead of a slipping layer), the simple concept of a step function in the fluidity is generally accepted as convention for the analysis of electrokinetic phenomena.
potential is identified as the potential at the supposed slipping plane that separates the stationary and the mobile phases in tangential flow of the liquid with respect to the surface. Although there is still very little direct evidence to support the view of a slipping plane (instead of a slipping layer), the simple concept of a step function in the fluidity is generally accepted as convention for the analysis of electrokinetic phenomena.
In general, it is believed that the plane of shear is located in the diffuse part of the double layer, i.e. adjacent to the Stern plane at the boundary of the Stern layer. This means that the potential is, in principle, lower than or at best equal to the potential at the onset of the diffuse double layer, . At low ionic strength the decay of the electrostatic potential as a function of the distance perpendicular to the surface is weak, under such circumstances it seems reasonable to assume that
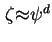 .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... criticism40
- (2001) Common practice is to use the bulk values of
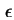 and
and 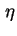 for the region beyond the Stern plane. However, it is imperative to indicate any assumptions regarding
and within the Stern layer.
for the region beyond the Stern plane. However, it is imperative to indicate any assumptions regarding
and within the Stern layer.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... potential41
- (2001) When it is assumed that
and have bulk values beyond the plane of sheer, one of the main remaining difficulties with the calculation of the potential is the role of the surface (excess) conductivity due to the excess charge in the double layer. Classical models, such as the Helmholtz-Smoluchowski theory of electrokinetic phenomena, neglect surface conductivity entirely. Once the potential is calculated, it is also possible to calculate the electrokinetic charge density,
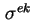 , with a suitable double layer model. By comparing
with
, with a suitable double layer model. By comparing
with
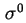 an impression can be gained about the amount of charge in the stagnant layer. General experience shows that often
an impression can be gained about the amount of charge in the stagnant layer. General experience shows that often
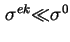 . Under the assumption that
, it follows that
. Under the assumption that
, it follows that
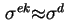 .
.
Notwithstanding the ambiguity in its determination, potentials play an important role in colloid science. Often electrokinetic measurements are the only available techniques to obtain information on the electrical potential near the surface.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... surface42
- cf. footnote to section 1.1.9
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... mass)43
- The notions 'specific surface area' and 'average area per molecule in the surface' are rarely used both in the same context, but if this should happen, it is particularly important to indicate clearly, which choice of symbols has been made.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.