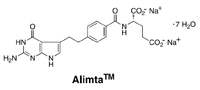
|
|
Vol.
33 No. 5
September-October 2011
by Edward C. Taylor
The following essay describes in broad terms the history of the discovery of Alimta. It was written as a part of a brochure celebrating the dedication of Princeton’s magnificent new chemistry building, completed by the end of 2010. The building, recognized as perhaps the finest, best-equipped, and designed facility for academic chemistry research in the country, was financed by royalties to Princeton from sales of Alimta by Eli Lilly & Co., to whom Princeton had given an exclusive license. The article was addressed to a general audience; my lecture in Glasgow1 filled in the organic and heterocyclic chemistry involved in the extensive explorations that finally led to the discovery and synthesis of Alimta.
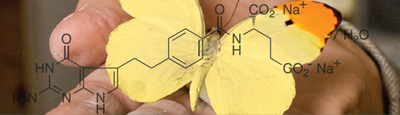 |
I have often been asked how an interest in the lovely white, yellow, and red pigments in the wings of butterflies could have led to the discovery of Alimta. For an answer, we have to go back 64 years to the fall of 1946, when I entered graduate school at Cornell University to major in organic chemistry. In searching for a possible thesis topic, I ran across an article in Science describing the isolation, properties, and structure of a strange compound that the researchers from the pharmaceutical company Lederle had isolated from human liver. It had the totally unexpected property of being necessary for the growth of a number of microorganisms. During their structural investigations of this compound, they found that its core was made up of two fused six-membered rings that constituted six carbon atoms and four nitrogen atoms, a core that had only been observed before as pigments in the wings of butterflies, and in the skins of tropical fish.
Shortly before I began my scientific studies, Robert Purrmann, a young organic chemist at the University of Munich, had taken up a problem that had frustrated some of the best organic chemists in Europe over a period of 50 years. This was the elucidation of the structures of the white and yellow pigments in butterfly wings. Purrmann finally managed to identify and subsequently synthesize both the white pigment of the white cabbage butterfly and the gorgeous yellow pigment of the brimstone butterfly, and he published these results in Liebig’s Annalen der Chemie in 1941.
Purrmann’s paper came to the attention of the Lederle chemists who were struggling with the structure of the compound from human liver. To their amazement, the two-ring core of the Lederle compound was almost identical to the butterfly wing pigments described by Purrmann. This coincidence of structure between the butterfly wing pigments and the core of the liver compound that was essential for the growth of bacteria struck me as stunningly bizarre, and I decided at that moment to devote my Ph.D. thesis to a study of the so-called pterin heterocyclic core and related heterocycles—where they come from in nature, their properties, both physical and biological, and methods for their preparation.
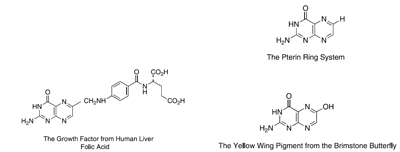 |
Upper right: pterin core that I studied as a part of my Ph.D. work.
Lower right: structure described in Purrmann’s paper in 1941.
Left: compound isolated from liver by Lederle scientists and reported in Science. |
I therefore spent the next three years working with these fascinating, frustrating compounds. It was abundantly obvious from the beginning of this project why it had taken more than 50 years before the structure of even the simplest of the butterfly wing pigments could be elucidated, for these compounds were not easy to work with. Their solubility, for example, rivaled that of Vermont granite, so that spectroscopic studies were difficult, and combustion analysis to determine molecular composition was seldom reliable because of the reluctance of these compounds to burn. (There was even a patent granted at that time for the use of related compounds as furnace liners.)
As it turned out, my choice of this area of organic chemistry for study proved to be extremely fortunate, for it led to a lifelong fascination with heterocyclic chemistry, and proved to be an invaluable training in the techniques of working in this experimentally challenging field.
At the same time that I was pursuing my thesis work at Cornell, the scientists at Lederle Laboratories reasoned, on the basis of the antimetabolite theory that was at that time a guiding principle of drug research, that it might be possible to develop a new group of antibacterial agents. If the structure of the liver compound could be modified, they hoped that bacteria might treat the modified compounds as if they were essential for its growth. Bacterial growth, however, should actually be inhibited because the modified structure would not function in the same way as the original. So Lederle made a change in the structure of the liver compound by substituting one nitrogen atom for an oxygen atom to give a compound called aminopterin, and replacing a hydrogen by a methyl group in the latter compound to give methotrexate. These new compounds indeed proved to be powerful antibacterial agents.
The compound isolated from human liver was shown to be the same substance that, albeit not identified structurally, had earlier been found in various leafy vegetables such as spinach, and was therefore named “folic acid” in 1941.
In 1948, it was found by others that aminopterin and methotrexate were effective in bringing about remissions in acute lymphoblastic leukemia in children. This startling revelation of the antitumor effects of blocking the action of folic acid immediately excited world-wide interest, for both the antibacterial and the antitumor activity of aminopterin and methotrexate were shown to be due primarily to blocking the action of a specific enzyme dihydrofolic acid reductase (DHFR) essential for the ultimate biosynthesis of both DNA and RNA in cells. These discoveries are considered to be the beginning of modern cancer chemotherapy.
A fundamental problem associated with this approach to cancer chemotherapy was the lack of specificity (for cancer cells rather than normal cells) of any of the DHFR inhibitors thus far discovered, and this held true for a number of newer DHFR inhibitors that were found during the following several decades. During this time, however, much was learned about the multiple roles of folate-derived coenzymes in cellular metabolism, and it became increasingly clear that a better approach might be to search for inhibitors of some of the other folate-dependent enzymes involved in other aspects of cellular biosynthetic processes. By this time some 20 such processes had been identified. One indication that this was a fertile area for potential drug development was the discovery in 1957 of 5-fluorouracil (5-FU), which blocked an enzyme known as thymidylate synthase that is key to the biosynthesis of DNA. Further insights included the discovery of the essential role of a different enzyme that catalyzes the conversion of monoglutamates such as folic acid itself, as well as aminopterin and methotrexate, to polyglutamates.
My own role in these decades-long developments from the 1946–48 discoveries was minor. After receiving my Ph.D. from Cornell, I spent a year in Zürich, Switzerland studying with Nobelist Professor Leopold Ruzicka under a National Academy of Sciences postdoctoral fellowship. In 1950, I studied at the University of Illinois as a postdoctoral fellow, where I was appointed to the faculty in 1951. I accepted a position at Princeton in 1954, and have been there ever since. Although I retained my fascination with the folic acid arena, and continued to work with the heterocyclic ring systems that had so fascinated me in graduate school, I spent most of the following several decades enjoying myself hugely in explorations of many diverse areas of chemistry such as photochemistry, organometallic chemistry, synthetic methodology, and the total synthesis of natural products, most of them quite unrelated to butterfly wing pigments.
It was not until the middle 1970s that I returned to cancer chemotherapy as a specific research goal. The incentive was the accumulation by others of much additional information on the complex pathways by which drugs such as methotrexate enter the cell, undergo activation and deactivation, and can be retained in or eliminated from the cell, as well as an understanding of the other multiple roles played by folate-dependent enzymes.
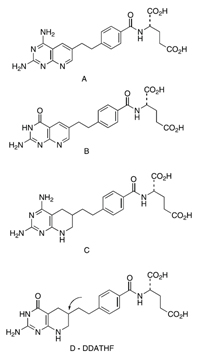
One of my first major new projects in this field involved the synthesis of 5,10-dideazaaminopterin (A), 5,10-dideazafolic acid (B), and the corresponding tetrahydro derivatives C and D (see figure to right). Compound D, 5,10-dideazatetrahydrofolic acid, subsequently became known by its acronym DDATHF. The deletion of two of the nitrogen atoms of aminopterin, folic acid, and their tetrahydro derivatives would be expected both to lower basicity and increase lipophilicity, and we therefore anticipated that these compounds might have different biological properties as a result. Of particular potential interest was D because, as a variant of tetrahydrofolic acid and not aminopterin, it might satisfy the requirements of a new type of inhibitor. For one thing, this new inhibitor would have to be accepted by the cell as a “normal substrate” for all of the transport and activation processes that were now understood to be essential for inhibitory activity. In addition, it had to function as a pseudo substrate for the enzymes involved in one-carbon atom transfers in cellular metabolism. Its structure, however, precluded its possible function as a one-carbon acceptor; i.e., it could not participate in any of the one-carbon transfer reactions known to be critical for the syntheses, inter alia, of purines and thymidylate, and therefore both DNA and RNA.
At this time, I had been for many years a consultant to both the medicinal chemistry and the process development programs at Eli Lilly & Co. in Indianapolis, Indiana. As a consequence, we knew each other well, and they readily agreed to subject these new potential antitumor candidates to initial evaluation. So I sent samples of these compounds to them, and crossed my fingers.
The first response I received was a curious one. Lilly reported that something appeared to be amiss with their tests, and they asked if I could send them an additional sample of D. Fortunately, there was a bit more, but not much more, of the compound, and I sent it to Lilly.
 |
Their next response, however, was a very excited apology. It appeared that nothing had been wrong with the first tests, but Lilly could not believe them and assumed that the tests were faulty. My compound D was one of the most active antitumor agents they had ever seen, and brought about 95–100 percent inhibition of growth in a broad range of tumors in mice and in human tumor xenografts. In addition, it was fully active against tumors that were resistant to methotrexate, indicating that it was acting to block tumor growth by a totally different mechanism. Further examination of this compound by Lilly showed convincingly that it killed tumor cells by blocking de novo purine biosynthesis within cells, thus preventing DNA and RNA biosynthesis.
It was clear that we had an extraordinarily exciting compound on our hands, and that we might be on the brink of a major breakthrough. Princeton does not have a medical school and thus could not provide a base for various aspects of the drug development process, let alone for clinical trials. We needed an active collaboration with a partner with extensive experience in drug development and with an active cancer program in place. A Princeton/Lilly collaboration was a natural one to investigate this exciting lead, and this was quickly agreed upon. I was to head up the chemistry effort, while Lilly took on the basic biochemical and pharmacological studies that are so critical for guiding the synthetic work, as well as the overall organization of this collaborative effort.
It seemed highly unlikely that DDATHF (compound D), whose extraordinary activity served to launch this collaborative program, would prove to be the ultimate structure of choice. We therefore embarked on what amounted to a classical SAR, or structure-activity-relationship study, where structural changes are made systematically, and each new compound emerging from this work is then evaluated. Every portion of the structure of DDATHF was probed in the hope that a picture of an optimal structure might emerge. We had to bear in mind that every change in structure can affect solubility, absorption, bioavailability, metabolism, partition coefficients, toxicity, possible activity against other biological targets, stability (pharmacokinetic and biochemical properties), as well as resonance, inductive effects, electronic distribution, chemical reactivity and stability, shape, size and bond angles, pKa and hydrogen bonding capacity (chemical properties). Furthermore, the effect of a change in one part of the molecule can modify or cancel what might have appeared to be a potentially useful change in another part of the molecule.
For me, this program proved to be a veritable playground for explorations of heterocyclic chemistry, since it called for the synthesis of many new ring systems and required the discovery of many new chemical transformations. Over the next four years, at least 800 new antitumor candidates were designed and synthesized, at Princeton and at Lilly, and each compound had to be thoroughly evaluated. To our astonishment, DDATHF itself proved to be one of the two most active compounds, another being a variation of DDATHF in which a thiophene ring replaced the benzene ring in the bridge region (see structure D).
| . . . to our amazement and delight, this new compound proved to be a veritable bombshell when it was evaluated at Lilly. |
One unavoidable step in the synthesis of both of these compounds was reduction of a substituted pyridine ring. As we had carried it out, the hydrogen addition was not stereospecific and resulted in the formation of two diastereomers. Because we wanted to evaluate the pharmacological properties of the individual diastereomers, the two diastereomers of DDATHF (and of the second, thiophene-containing candidate) had either to be separated and evaluated separately, or individually synthesized. In discussions with Lilly, it was decided that Lilly would undertake the separation challenge, while Princeton would tackle the synthesis challenge.
At Lilly, my principal chemical collaborator, Dr. Joe Shih, succeeded in the separation of the two diastereomers of DDATHF. The yields were extremely low: diastereomer 1 was obtained only in 3–5 percent yield, and diastereomer 2 was obtained in 15 percent yield. The latter was chosen to be the clinical candidate and was named lometrexol. Some time later, the two diastereomers of the thiophene-containing candidate were also separated by Dr. Shih, and one of the two, named LY309887, was also chosen for clinical trial. However, preparing sufficient quantities of these materials for clinical trials appeared to be a herculean task, since the reduction step, followed by the mandatory separation of diastereomers (in very low yield), occurred late in a multistep total synthesis. Although the single diastereomer 2 indeed went into clinical trial, an alternative approach to its preparation was a high priority.
While Lilly was struggling with the separation problems, I tried at Princeton one approach after another to devise a stereospecific strategy for the preparation of diastereomer 2 alone. After many, many failures, I decided to dodge the problem entirely by designing a new candidate that could not exist in two diastereomeric forms. This approach required the design of a compound that did not possess the stereogenic carbon atom that was responsible for the two diastereomers resulting from the pyridine reduction step. To accomplish this, I focused on a new ring system that retained as many of the structural features of DDATHF as possible so I could assess the impact of the new ring system on activity. However, there was no way one could predict the consequence of what amounted to a very significant change in the structure of DDATHF, and the abandonment of some of the structural features that we had already determined to be critically important for activity.
Certain fragments of our projected new target compound were already in hand, and this facilitated the design and implementation of its synthesis. Dr. Dietmar Kuhnt joined my group at that time as a postdoc, and I asked him to prepare the compound. And to our amazement and delight, this new compound proved to be a veritable bombshell when it was evaluated at Lilly.
After extensive attempts by both Princeton and Lilly to improve on this compound, we found that this was still the most active compound in the series, and it was chosen for clinical trials. This compound is now known as Alimta.
Most striking was its remarkable activity against thymidylate synthase (TS). This folate-dependent enzyme is required for the biosynthesis of thymidine, and thus DNA, and finding an effective inhibitor of TS was considered at that time to be the Holy Grail for cancer researchers. Furthermore, our new compound also proved to be an inhibitor of several other folate dependent enzymes including dihydrofolate reductase and glycinamide ribonucleotide N-formyltransferase. It was therefore, in essence, and all by itself, the equivalent of a cocktail of antitumor agents, with the advantages of the usual antitumor cocktail (a combination of different drugs with different targets of action) but without its disadvantages (a combination of the toxicities of each drug in the mixture).
The transformation of Princeton’s new clinical candidate compound into an effective and successful cancer drug has been described as possessing all of the elements of a great action movie – overcoming obstacles, detective work, perseverance and, ultimately, a triumph for patients. It was the result of the remarkable scientific collaboration between Princeton and Lilly that should stand as a model of what can be possible when academia and industry truly work together.
Everyone was ecstatic when early clinical trials showed that almost all solid tumors examined responded to Alimta. Perhaps the most remarkable of all the responses was that with malignant pleural mesothelioma, the devastating lung cancer caused primarily by exposure to asbestos. No clinically effective treatment for mesothelioma was previously known.
The results of clinical trials in mesothelioma were so promising that even before the drug was approved, the FDA allowed Lilly to provide it free of charge to more than 1000 medically eligible patients through a compassionate use program.
Many obstacles lay ahead. For instance, in the last phase of stability testing, glass particles appeared in the vials of Alimta. These particles were caused by a totally unexpected interaction of liquid Alimta and the glass. Lilly scientists quickly reconstituted the drug into a new powdered formulation. In addition, during a Phase I clinical trial, some patients reacted to Alimta administration with skin rash. Lilly found that this effect could be mitigated by the addition of a low dose of oral steroids to the treatment regimen.
In 2004, Alimta, in combination with cisplatin, received its first FDA approval for the treatment of malignant pleural mesothelioma when surgery was not an option. Within five years, Alimta received three more approvals for the treatment of advanced nonsquamous non-small cell lung cancer. Through these clinical trials, researchers have shown that tumor histology can be used to determine which NSCLC patients may benefit from Alimta treatment.
Today, Lilly is continuing studies of Alimta in combination with other therapies and in new tumor types at cancer centers around the world.
Edward C. Taylor <[email protected]> is A. Barton Hepburn Professor of Organic Chemistry Emeritus at Princeton University. Taylor’s career has involved pioneering studies in organic synthesis methodology, organometallic chemistry, natural products, and medicinal chemistry.
1. Glasgow was the site of the 23rd International Congress on Heterocyclic Chemistry held 31 July–4 August 2011. Other papers presented at the ICHC-23 will appear in the IUPAC journal Pure and Applied Chemistry.
Page
last modified 26 August 2011.
Copyright © 2003-2011 International Union of Pure and Applied Chemistry.
Questions regarding the website, please contact [email protected] |