|
|
Vol.
28 No. 1
January-February 2006
"Inner Chemical Life" of Solids
by Joachim Maier
A quick glance at our environment is enough for us to realize the significance of the solid state in our daily life. Solids are advantageous over other forms of matter mainly because of their rigidity: durable structuring is inconceivable without solid matter which is characterized by low diffusion coefficients, at least for one component. Nonetheless, other components may exhibit perceptible or even fast diffusion without letting the solid lose its structural stability. Such a mobility is not only important to enable electrochemical performances, it is also a prerequisite for solid-state reactions to occur. In materials in which all atomic constituents are immobile, the electrons still can be fairly mobile, which means that electronic transport can be tuned reversibly. Solids exhibiting both ionic and electronic mobility are important for specific applications as well. Hence, apart from mere mechanical functionality, solids offer the possibility of subtly and reproducibly tailoring chemical and electrochemical functionalities.
Reactions involving solids are used extensively in laboratory research. Despite this fact, the chemists’ relationship to solids resembles that of a stepmother with regard to an unloved child, and is often more characterized by tolerance than by understanding. The chemists learned to prepare solid compounds of great complexity and to understand in detail perfect structure and bonding. However, this conception of the solid state is static and superficial, and does not hold the key for the tailoring of properties. Nor does it enable the understanding of kinetic processes in solid-state materials. Solids are still considered to be more “dead than alive”; they are, within the range of their stability, considered as chemically invariant entities. Only the surface is recognized as a site of chemical reactivity. The conception that solids have an “inner chemical life,” which makes it possible for us to tune their properties like we do with liquids, still sounds adventurous to most chemists. On the other hand, many materials scientists, ceramicists, and even physicists appreciate the chemical tunability of solids more than chemists.
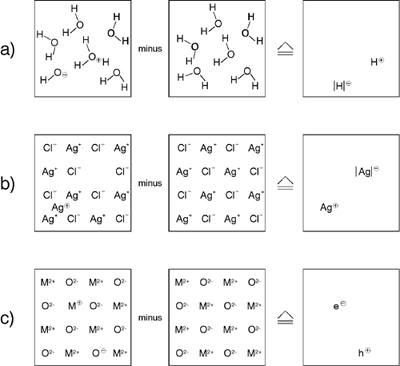
A comparison between the liquid and solid phases of water (figure 1) makes it immediately clear what is lacking in the naive chemical description of the solid state. As regards water, chemists are not so much interested in the bulk structure (in fact studying the detailed structure of water was largely the subject of physics), rather they are focusing on the deviations from the ideal water structure, namely the chemical excitations H3O+ and OH-. These species are, indeed, the acid-base active particles of relevance, and together with the dissolved species they determine the inherent acid-base and redox-chemistry. In addition, they are the relevant centers to be considered with respect to mass transport and electrical conductance. On the other hand, concerning the solid state (e.g., an ionic crystal such as AgCl), the historical situation is diametrically different. Consideration of the perfect solid-state structure was and still is the domain of chemistry while the genuine chemical issues such as mass transport, ion conduction, and the kinetics of compositional change are almost unaccounted for in the realm of chemistry. It is exactly this issue that prevents the solid state from being included in the familiar concept of chemical kinetics.
|
| Figure 1: a) As the compositionally unperturbed structure (chemical ground structure) is subtracted from the real structure, the point defects shown on the right remain. Naturally each is surrounded by a distorted region (effective radius of the point defect) which affects at least the immediate neighborhood. In the case of fluid phases (see above) this procedure can only be regarded as an instantaneous picture. (Owing to the absence of defined sites no distinction is made between various types of defect reactions as is done in the solid state.) b) Frenkel disorder is sketched in the second row. c) The third row shows the case of purely electronic disorder whereby localized charge carriers are assumed for the sake of clarity. Reprinted with permission from reference 1. |
This article emphasizes that it is the field of defect chemistry in solids, which represents the missing link in the conception.1 Defect chemistry not only provides the pertinent fundamental insight, it also enables optimization of solids with regard to technological applications. It should become implicitly clear that the physico-chemical consideration of the “mixed conductor,” which exhibits ionic and electronic mobility, allows generalization of semiconductor physics, solid-state ionics and electrochemistry.
The key to the “opening” of the solid, which in fact was provided as early as the 1930s by Frenkel and in particular by the physicist Schottky and the chemist Wagner,2 is the identification of the relevant centers in solids which are analogous to H3O+ and OH- in water. These are the so-called point defects. This is easy to demonstrate. Just remove from the dissociation
equation
(1)
or more concisely from
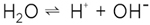 (2) (2)
an additional water molecule, as it is displayed in figure 1, then obviously we are left with two defects, viz. the excess proton H+ and the proton vacancy |H|-
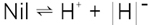 (3) (3)
The same procedure is possible with an ionic crystal such as AgCl. As above, it is entropy not energy that favors a disorder, as shown in figure 2. To a certain extent, excess Ag+ ions (interstitial ions) are formed at the expense of (Ag+) vacancies:
This Ag+ transfer may also be written as
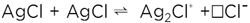 (4) (4)
(Owing to the rigidity of the lattice we have—in contrast to reaction (1)—to consider the vacancy ( ) explicitly.) ) explicitly.)
Most concisely (subtraction of two AgCl) it reads
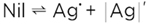 (5) (5)
This is not a mere formalism, in fact the perfect AgCl crystal, even though AgCl is dissociated into Ag+ and Cl-, is not dissociated in terms of being split into free particles. In the perfect state, the Ag+ and Cl- ions are strongly bound and can only vibrate around the lattice positions. In this sense equation (5) describes the “superionic” dissociation. Only this thermal disorder (together with dopants and neighboring phase effects) leads to mobile particles. In addition, the particles Ag· and |Ag|' (the old-fashioned symbols · and ' are used to highlight the significance of the relative charge they possess), formed for entropic reasons, not only exhibit an appreciable mobility, they also exhibit a higher local energy, and thus a higher reactivity that unfolds when they are in contact with reaction partners (e.g., at the surface).1
|
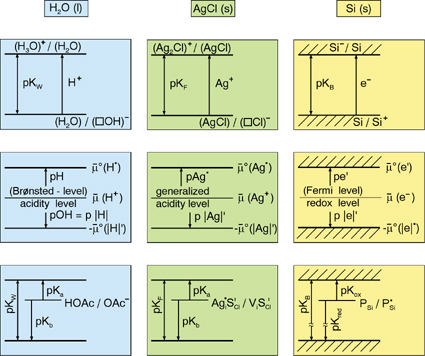
| Figure 2: In the same way as the concentration of protonic charge carriers characterizes the acidity (basicity) of water and in the same way as the electronic charge carriers characterize the redox activity, the concentration of elementary ionic charge carriers, that is, of point defects, measure the acidity (basicity) of ionic solids, while associates constitute internal acids and bases. The definition of acidity/basicity from the (electro-)chemical potential of the exchangeable ion, and, hence, of the defects leads to a generalized and thermodynamically firm acid-base concept that also allows to link acid-base scales of different solids (In order to match the decadic scale the levels are normalized by ln 10).2 Reprinted with permission from reference 3. |
In fact, the correspondence is so complete (figure 2) that one can show that a thermodynamically firm acid-base concept can (and in fact shall) be based on counting the equilibrium point defects in the same way that water pH and pOH reflect acidity and basicity in H2O.3
Together with the electronic carriers, the ionic defects constitute the internal acid-base and redox chemistry. Analogously to the ionic disorder described by equations (4) or (5), the electronic disorder is described by
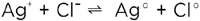 (6) (6)
or more concisely (subtracting Ag+Cl-- on both sides) by
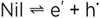 (7) (7)
(Equation 7 is to be preferred since it avoids double counting and is independent of the band structure. In AgCl the valence and conduction bands are only approximately due to Cl-p-orbitals and Ag-d-orbitals only.)
The presence of defects also allows the interaction with the neighboring phase. Every solid phase possesses a finite phase width. Even if the variability of the stoichiometry in MO1+δ may be so small that it does not perceptibly affect the total free energy, its influence on the defect budget and hence on all the parameters that directly rely on the presence of the charge carriers (conductivity, reactivity), is of first order. So the equilibration under different oxygen partial pressures can change normal conductors into superconductors, n-type conductors into p-type conductors, or ion conductors into electron conductors. Dopants in solids are comparable to, and are in fact as important as, impurities in water (e.g., consider traces of HCl in H2O with respect to pH or conductivity, see figure 2). Whilst in water both impurity cation and impurity anion are usually soluble, in solids most impurities are introduced substitutionally. Hence, the charge difference between dopant and substituted particle matters significantly. In fact, this effective charge is the only quantity that has to be known in most examples in order to predict the consequence of doping: a substitutional doping by a lower-valent cation, for example, introduces a negative relative-excess charge resulting in an enhanced concentration of positively charged carriers (holes, oxygen vacancies) and a depression of negatively charged carriers (e.g., conduction electrons, oxygen interstitials). It is therefore expected that Gd doping of CeO2 turns CeO2 from an electronic into an ionic conductor, and that Sr doping in La2CuO4 (Sr2+ replaces La3+) increases p-conduction (conduction via h·). Simple internal mass-action laws even show in a straightforward way that the concentration of hole pairs 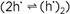 has to increase in the latter case. To explain, however, their amount or the fact that the formation of these so-called Cooper pairs enable superconductivity at finite temperatures requires a subtle understanding of the underlying quantum mechanics. has to increase in the latter case. To explain, however, their amount or the fact that the formation of these so-called Cooper pairs enable superconductivity at finite temperatures requires a subtle understanding of the underlying quantum mechanics.
In addition, point defects can internally react with each other: such association between ionic centers represents internal acids or bases (see figure 2, bottom center), while association of ionic with electronic particles represents internal redox centers (see figure 2, bottom right hand side); association between electronic carriers are of no less importance, examples are excitons (e'h·) or the already mentioned Cooper pairs ((e')2, (h·)2).
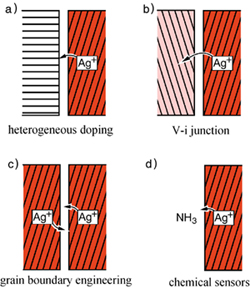
A further point that is equally important and is of significance in solid-state science is the influence of interfaces on the charge-carrier density.4 As in the case of solid/liquid interfaces, double layers at solid-solid surfaces result in an enormous change in concentration compared to the bulk values. It can be shown that based upon this concept new electrolytes or novel sensors can be created. This is particularly of importance when considering a high density of interfaces as is the case in nano-sized systems5 (see figure 3).
|
| Figure 3: Four basic space charge situations involving ionic conductors (here silver ion conductor): a) contact with an insulator that adsorbs Ag+ leads to an increased vacancy concentration adjacent, b) contact with a second ion conductor leads to a redistribution over two space charge layers, c) grain boundary that traps silver ions increases vacancy concentrations on both sides, d) contact with a fluid phase; the increase of the vacancy concentration if NH3 is adsorbed can be used as a selective sensor signal.4 Reprinted from J. Maier, in Modern Aspects of Electrochemistry, (B. E. Conway, ed.), vol. 38 (2005) 1, with permission from Springer, New York. |
In the extreme case of minuscule interfacial spacings, the interfaces begin to perceive each other and we are met with size effects. The field of nano-ionics, which is concerned with size effects on ionic transport properties, is expected to play a similar role for electrochemical applications as nano-electronics does for semiconductor devices.5
A further key point is chemical kinetics. The major difference to solution chemistry lies in the heterogeneity of the reactions. They start at the surface (e.g., at the gas/solid contact) and are followed by transport steps. Realizing that the hopping process in diffusion kinetics (hopping from one site to the next equivalent one) just refers to the simplest possible chemical kinetic problem, namely a “reaction” involving a symmetrical reaction profile (i.e., equal rate constants for forward and backward reactions), may remove a further hindrance for chemists to enter the field.1,6
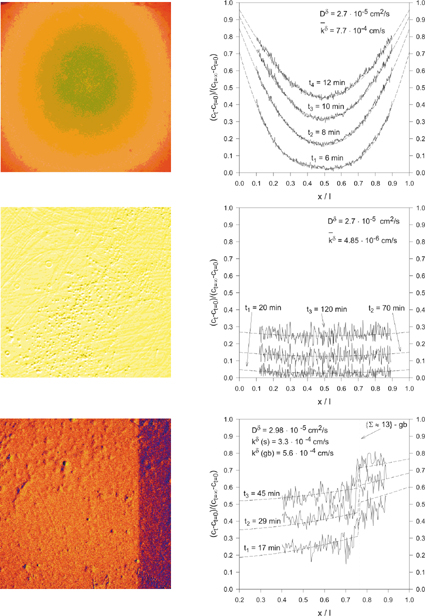
Figure 4 shows how, using the example of oxygen incorporation into an oxide, the variation of the stoichiometry proceeds. On changing the outer oxygen partial pressure, three basic kinetic modes for the kinetics of stoichiometry change in a perovskite (SrTiO3) can occur as displayed.
Defect chemistry is not only a prerequisite for adequately understanding the fundamental aspects of the internal chemistry of solids or the proper understanding of catalysis, it is also a prerequisite for enabling functionality and designability of solids, as far as many important applications are concerned: Oxygen ion or proton conductors are the key component of high-temperature fuel cells (SOFC); oxide electrodes that may incorporate oxygen or lithium are relevant electrodes in SOFCs and Li batteries, respectively. The latter are mixed conductors and are also relevant for permeation membranes or catalysts. Also, gas sensors rely on the interaction of solids with the neighboring gas phase mediated by point defects. (Figure 5 gives an overview of such devices.) As far as the engineering of relevant functional materials is concerned, tuning of ionic and electronic conductivities is a key issue
Even if one is exclusively concerned with semiconductor devices, stoichiometric aspects are crucial. Defect chemistry offers the possibility to understand and predict the fine composition in the frozen-in state that is otherwise only accessible in terms of empirical preparation procedures or via an a posteriori analysis.7
These are only a few examples out of the whole spectrum of relevant applications, but they may suffice to show (i) that the internal chemistry of solids is conceptually a viable subset of chemistry, (ii) how efficiently electrochemical and kinetic properties can be purposefully tuned given a pertinent understanding of this topic, and (iii) how subtly the understanding of electroceramic applications is connected with defect chemistry.
Joachim Maier is a professor and director at the Max Planck Institut fuer Festkoerperforschung in Stuttgart, Germany. He has also been a titular member of the IUPAC Physical and Biophysical Chemistry Division.
References
1 J. Maier, Physical Chemistry of Ionic Materials. Ions and Electrons in Solids, John Wiley & Sons, Chichester, 2004.
2 C. Wagner, W. Schottky, Z. Phys. Chem. B11 (1930) 163; C. Wagner, Z. Phys. Chem. B32 (1936) 447.
3 J. Maier, Chem. Eur. J. 7 (22) (2001) 4762.
4 J. Maier , Prog. Solid St. Chem. Vol. 23 (3) (1995) 171.
5 J. Maier, Nature Materials 4 (11) (2005) 805.
6 H. Schmalzried, Chemical Kinetics of Solids, VCH, Weinheim, 1995.
7 J. Maier, Phys. Chem. Chem. Phys. 5 (11) (2003) 2164.
|
| Figure 4: Three limiting cases of oxygen indiffusion into SrTiO3 as recorded by spatially resolved in-situ spectroscopy. L.h.s.: snapshots during the experiment. R.h.s.: The evolution of the corresponding stoichiometry profiles. Top: diffusion control. Centre: Surface reaction control. Bottom: Grain Boundary control. Reprinted from J. Maier, Solid State Ionics 135 (1-4) (2000) 575, with permission from Elsevier. |
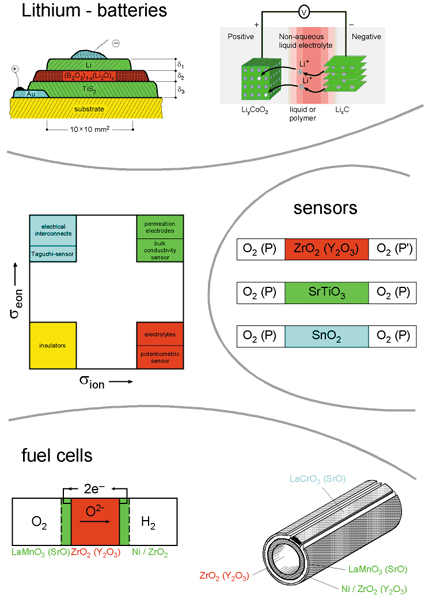 |
Figure 5: Overview of typical electrochemical devices. The key components are materials of different ratios of ionic and electronic conductivities. Reprinted from J. Maier, Radiat. Eff. Defects Solids 158 (2003) 1, with permission from Taylor & Francis. |
Page
last modified 27 December 2005.
Copyright © 2003-2006 International Union of Pure and
Applied Chemistry.
Questions regarding the website, please contact [email protected]
|